Brenda Klemm Arci Mattos de Freitas Alves, Alexandra Prufer de Queiroz Campos Araujo, Flávia Nardes Dos Santos, Márcia Gonçalves Ribeiro
{"title":"Type-1 spinal muscular atrophy cohort before and after disease-modifying therapies.","authors":"Brenda Klemm Arci Mattos de Freitas Alves, Alexandra Prufer de Queiroz Campos Araujo, Flávia Nardes Dos Santos, Márcia Gonçalves Ribeiro","doi":"10.1055/s-0044-1791757","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong> Spinal muscular atrophy (SMA-5q) is a neurodegenerative disease characterized by progressive muscle atrophy, hypotonia, and weakness, with SMA 1 presenting symptoms within the first 6 months of life. Disease-modifying therapies have been approved, with better outcomes with earlier treatment.</p><p><strong>Objective: </strong> To describe the safety and clinical efficacy of disease-modifying therapies based on <i>SMN1</i> and <i>SMN2</i> gene strategies concerning motor, respiratory, and bulbar function. Patients with SMA 1 were divided into 2 groups: those exclusively on nusinersen (group 1) and those transitioning to onasemnogene abeparvovec (OA) (group 2).</p><p><strong>Methods: </strong> Over 18 months, patients were assessed using the Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP-INTEND) scale, developmental milestones, ventilation needs and duration, nutritional support needs, consistency of food, and signs of dysphagia. There were ten patients, divided between the groups; in group 1, the average age for starting nusinersen was 53.6 (12-115) months, and, in group 2, the age was 7 (1-12) months for nusinersen and 15.2 (10-19) months for OA.</p><p><strong>Results: </strong> Our results indicate that 70% of patients reached some motor milestones, with group 1 increasing by 10.2 points on the CHOP-INTEND scale, while group 2 increased by 33 points. Additionally, 90% of the patients experienced no respiratory decline, and 30% maintained oral feeding. No serious adverse effects or deaths were recorded.</p><p><strong>Conclusion: </strong> Both groups showed improvement in motor function and stabilization of respiratory and bulbar function, with the difference between the groups possibly being related to the earlier treatment initiation. Thus, the present study provides valuable insights into the real-world safety and clinical efficacy of disease-modifying therapies for SMA 1 patients.</p>","PeriodicalId":8694,"journal":{"name":"Arquivos de neuro-psiquiatria","volume":"82 11","pages":"1-8"},"PeriodicalIF":1.6000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11540468/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Arquivos de neuro-psiquiatria","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1055/s-0044-1791757","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/6 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"NEUROSCIENCES","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Spinal muscular atrophy (SMA-5q) is a neurodegenerative disease characterized by progressive muscle atrophy, hypotonia, and weakness, with SMA 1 presenting symptoms within the first 6 months of life. Disease-modifying therapies have been approved, with better outcomes with earlier treatment.
Objective: To describe the safety and clinical efficacy of disease-modifying therapies based on SMN1 and SMN2 gene strategies concerning motor, respiratory, and bulbar function. Patients with SMA 1 were divided into 2 groups: those exclusively on nusinersen (group 1) and those transitioning to onasemnogene abeparvovec (OA) (group 2).
Methods: Over 18 months, patients were assessed using the Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP-INTEND) scale, developmental milestones, ventilation needs and duration, nutritional support needs, consistency of food, and signs of dysphagia. There were ten patients, divided between the groups; in group 1, the average age for starting nusinersen was 53.6 (12-115) months, and, in group 2, the age was 7 (1-12) months for nusinersen and 15.2 (10-19) months for OA.
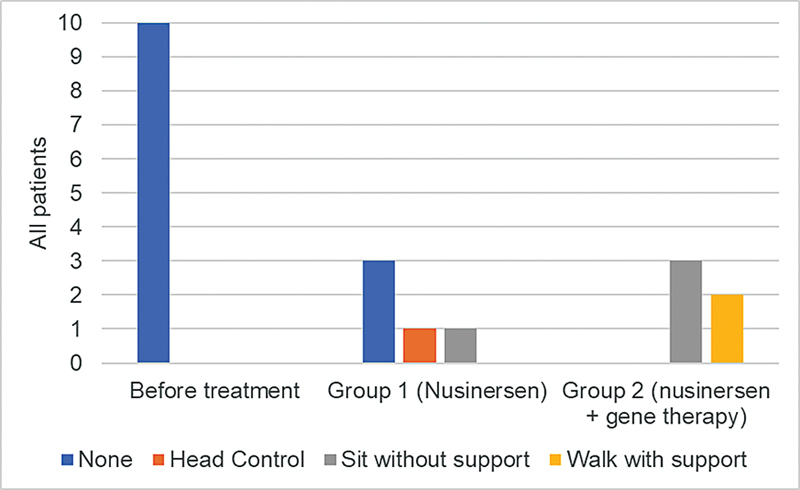
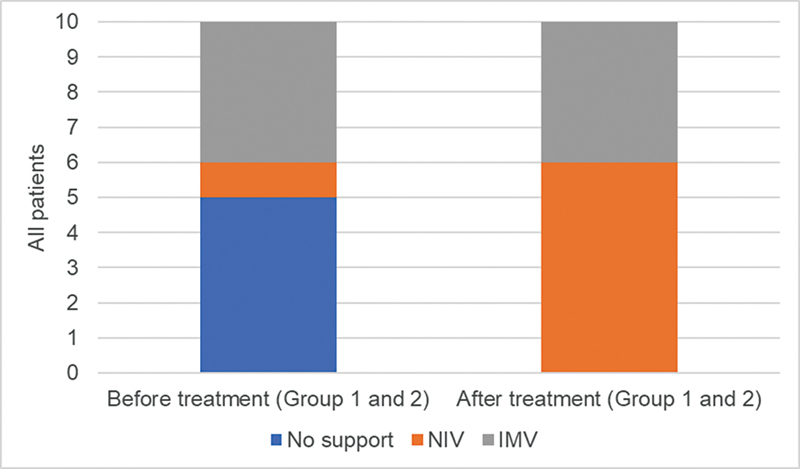
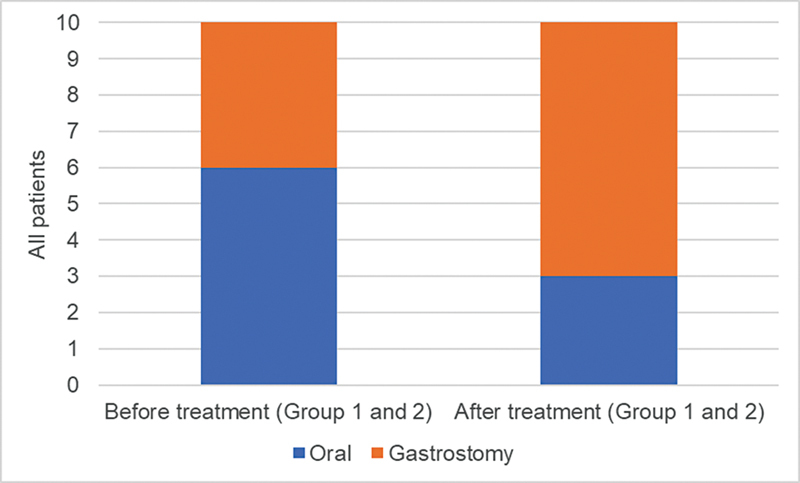
Results: Our results indicate that 70% of patients reached some motor milestones, with group 1 increasing by 10.2 points on the CHOP-INTEND scale, while group 2 increased by 33 points. Additionally, 90% of the patients experienced no respiratory decline, and 30% maintained oral feeding. No serious adverse effects or deaths were recorded.
Conclusion: Both groups showed improvement in motor function and stabilization of respiratory and bulbar function, with the difference between the groups possibly being related to the earlier treatment initiation. Thus, the present study provides valuable insights into the real-world safety and clinical efficacy of disease-modifying therapies for SMA 1 patients.
期刊介绍:
Arquivos de Neuro-Psiquiatria is the official journal of the Brazilian Academy of Neurology. The mission of the journal is to provide neurologists, specialists and researchers in Neurology and related fields with open access to original articles (clinical and translational research), editorials, reviews, historical papers, neuroimages and letters about published manuscripts. It also publishes the consensus and guidelines on Neurology, as well as educational and scientific material from the different scientific departments of the Brazilian Academy of Neurology.
The ultimate goals of the journal are to contribute to advance knowledge in the areas of Neurology and Neuroscience, and to provide valuable material for training and continuing education for neurologists and other health professionals working in the area. These goals might contribute to improving care for patients with neurological diseases. We aim to be the best Neuroscience journal in Latin America within the peer review system.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


