{"title":"Variable Mechanisms for Cobalt-Catalyzed Alkyne Dimerization Pinpointed by Quasi-Classical Trajectory Simulations","authors":"Haohua Chen, Zhile Dang, Xiantong Sha, Yu Wang, Zhiguo Zhang, Yixin Luo* and Yu Lan*, ","doi":"10.1021/acscatal.4c0374910.1021/acscatal.4c03749","DOIUrl":null,"url":null,"abstract":"<p >Transition metal-catalyzed alkyne dimerization represents a powerful method for the construction of enynes. However, the ambiguous hydrogen transfer mechanism during the dimerization has resulted in controlling the regio-, stereo-, and, where applicable, chemoselectivity remaining a long-standing challenge. Herein, a combination of DFT calculations and quasi-classical MD simulations was used to interrogate the dynamic motion of hydrogen in cobalt-catalyzed alkyne dimerization. The collective results inspired us to propose, for the first time, a substrate-dependent differential hydride transfer model involving either concerted oxidative hydrogen transfer or stepwise oxidative addition, followed by alkyne insertion. The practicability and universality of this oxidative hydride transfer mechanism were further validated by the theoretical studies of experimentally observed selective cross- and homo-dimerization. Charge distribution analyses depicted that the differentiation between those two hydride transfer mechanisms originates from the α-silicon effect, which can stabilize the neighboring negative charge of the alkyne. Furthermore, a comprehensive DFT study of the substituent effects of alkynes reveals that the electron-withdrawing group will accelerate the oxidative hydride transfer process, which can open up avenues for mechanistic-oriented selective dimerization.</p>","PeriodicalId":9,"journal":{"name":"ACS Catalysis ","volume":"14 21","pages":"16469–16478 16469–16478"},"PeriodicalIF":13.1000,"publicationDate":"2024-10-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Catalysis ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acscatal.4c03749","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
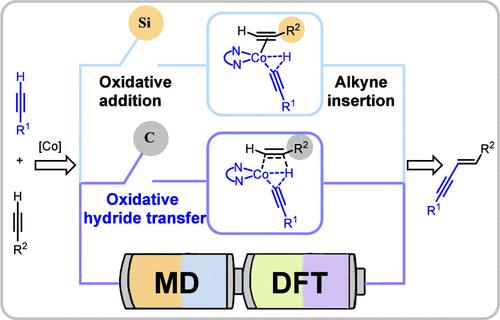
Abstract
Transition metal-catalyzed alkyne dimerization represents a powerful method for the construction of enynes. However, the ambiguous hydrogen transfer mechanism during the dimerization has resulted in controlling the regio-, stereo-, and, where applicable, chemoselectivity remaining a long-standing challenge. Herein, a combination of DFT calculations and quasi-classical MD simulations was used to interrogate the dynamic motion of hydrogen in cobalt-catalyzed alkyne dimerization. The collective results inspired us to propose, for the first time, a substrate-dependent differential hydride transfer model involving either concerted oxidative hydrogen transfer or stepwise oxidative addition, followed by alkyne insertion. The practicability and universality of this oxidative hydride transfer mechanism were further validated by the theoretical studies of experimentally observed selective cross- and homo-dimerization. Charge distribution analyses depicted that the differentiation between those two hydride transfer mechanisms originates from the α-silicon effect, which can stabilize the neighboring negative charge of the alkyne. Furthermore, a comprehensive DFT study of the substituent effects of alkynes reveals that the electron-withdrawing group will accelerate the oxidative hydride transfer process, which can open up avenues for mechanistic-oriented selective dimerization.
期刊介绍:
ACS Catalysis is an esteemed journal that publishes original research in the fields of heterogeneous catalysis, molecular catalysis, and biocatalysis. It offers broad coverage across diverse areas such as life sciences, organometallics and synthesis, photochemistry and electrochemistry, drug discovery and synthesis, materials science, environmental protection, polymer discovery and synthesis, and energy and fuels.
The scope of the journal is to showcase innovative work in various aspects of catalysis. This includes new reactions and novel synthetic approaches utilizing known catalysts, the discovery or modification of new catalysts, elucidation of catalytic mechanisms through cutting-edge investigations, practical enhancements of existing processes, as well as conceptual advances in the field. Contributions to ACS Catalysis can encompass both experimental and theoretical research focused on catalytic molecules, macromolecules, and materials that exhibit catalytic turnover.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


