Chemical shift prediction in 13C NMR spectroscopy using ensembles of message passing neural networks (MPNNs)
IF 2
3区 化学
Q3 BIOCHEMICAL RESEARCH METHODS
引用次数: 0
Abstract
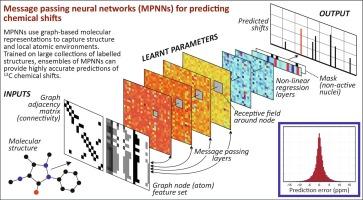
This study reports a deep learning approach that utilises message passing neural networks (MPNNs) for predicting chemical shifts in 13C NMR spectra of small molecules. MPNNs were trained on two distinct datasets: one with approximately 4000 labelled structures and another with over 40,000. To reduce stochastic variation, an ensemble framework was implemented, which is simple to deploy on multiple nodes of a High-Performance Computing facility.
The results emphasise the critical role of training set size and diversity. While prediction performance was comparable on test sets drawn from each dataset, the ensemble trained on the larger dataset retained its accuracy when these sets were crossed over, and when applied to a further collection of approximately 12,000 previously unseen structures introduced after all development work had been completed. In contrast, the ensemble trained on the smaller dataset showed a notable decline in generalisation ability. This difference is attributed to the greater diversity of atomic environments captured in the larger dataset.
The larger dataset also enabled more robust modelling of various error properties, providing a quantitative foundation for spectral assignment and verification. This was achieved in two ways. First, a clear relationship was observed between prediction errors and the frequency of different node feature vectors in the training data, allowing error estimates to be associated with individual nodes based on their type. These estimates can be used as weights in a modified cityblock distance metric when assigning observed to predicted shifts. Second, the mean absolute prediction error calculated at the structure level is well-fitted by a Gaussian kernel cumulative distribution. This enabled a probabilistic assessment of whether the predicted shifts and assigned observations are consistent with originating from the same molecular structure.
利用信息传递神经网络 (MPNN) 集合预测 13C NMR 光谱中的化学位移
本研究报告了一种利用消息传递神经网络(MPNN)预测小分子 13C NMR 光谱中化学位移的深度学习方法。MPNN 在两个不同的数据集上进行了训练:一个数据集包含约 4000 个标记结构,另一个数据集包含 40,000 多个标记结构。为了减少随机变化,我们实施了一个集合框架,该框架很容易部署在高性能计算设备的多个节点上。虽然从每个数据集抽取的测试集的预测性能相当,但在较大数据集上训练的集合在这些数据集交叉使用时,以及在所有开发工作完成后应用于约 12,000 个以前未见过的结构的进一步集合时,仍保持了其准确性。相比之下,在较小数据集上训练的集合的泛化能力明显下降。这种差异归因于较大数据集中捕捉到的原子环境更加多样化。较大数据集还能对各种误差属性进行更稳健的建模,为光谱分配和验证提供定量基础。这可以通过两种方式实现。首先,在预测误差与训练数据中不同节点特征向量的频率之间发现了明确的关系,从而可以根据节点类型将误差估计值与单个节点联系起来。在将观测到的偏移分配到预测的偏移时,这些估计值可用作修改后的城市街区距离度量的权重。其次,在结构层面计算出的平均绝对预测误差与高斯核累积分布拟合良好。这样就可以从概率上评估预测偏移和分配的观测值是否符合同一分子结构。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

CiteScore
3.80
自引率
13.60%
发文量
150
审稿时长
69 days
期刊介绍:
The Journal of Magnetic Resonance presents original technical and scientific papers in all aspects of magnetic resonance, including nuclear magnetic resonance spectroscopy (NMR) of solids and liquids, electron spin/paramagnetic resonance (EPR), in vivo magnetic resonance imaging (MRI) and spectroscopy (MRS), nuclear quadrupole resonance (NQR) and magnetic resonance phenomena at nearly zero fields or in combination with optics. The Journal''s main aims include deepening the physical principles underlying all these spectroscopies, publishing significant theoretical and experimental results leading to spectral and spatial progress in these areas, and opening new MR-based applications in chemistry, biology and medicine. The Journal also seeks descriptions of novel apparatuses, new experimental protocols, and new procedures of data analysis and interpretation - including computational and quantum-mechanical methods - capable of advancing MR spectroscopy and imaging.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


