Improving the reliability of classical molecular dynamics simulations in battery electrolyte design
IF 13.1
1区 化学
Q1 Energy
引用次数: 0
Abstract
Explorations into new electrolytes have highlighted the critical impact of solvation structure on battery performance. Classical molecular dynamics (CMD) using semi-empirical force fields has become an essential tool for simulating solvation structures. However, mainstream force fields often lack accuracy in describing strong ion-solvent interactions, causing disparities between CMD simulations and experimental observations. Although some empirical methods have been employed in some of the studies to address this issue, their effectiveness has been limited. Our CMD research, supported by quantum chemical calculations and experimental data, reveals that the solvation structure is influenced not only by the charge model but also by the polarization description. Previous empirical approaches that focused solely on adjusting ion-solvent interaction strengths overlooked the importance of polarization effects. Building on this insight, we propose integrating the Drude polarization model into mainstream force fields and verify its feasibility in carbonate, ether, and nitrile electrolytes. Our experimental results demonstrate that this approach significantly enhances the accuracy of CMD-simulated solvation structures. This work is expected to provide a more reliable CMD method for electrolyte design, shielding researchers from the pitfalls of erroneous simulation outcomes.
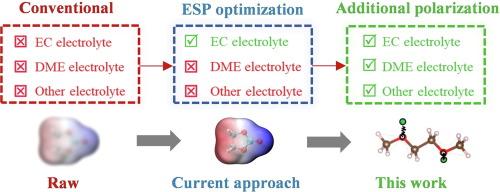
提高经典分子动力学模拟在电池电解质设计中的可靠性
对新型电解质的探索凸显了溶解结构对电池性能的重要影响。使用半经验力场的经典分子动力学(CMD)已成为模拟溶解结构的重要工具。然而,主流力场在描述离子与溶剂之间的强相互作用时往往缺乏准确性,导致 CMD 模拟与实验观察之间存在差异。虽然一些研究采用了一些经验方法来解决这一问题,但其效果有限。我们的 CMD 研究在量子化学计算和实验数据的支持下发现,溶解结构不仅受电荷模型的影响,还受极化描述的影响。以前的经验方法只注重调整离子与溶剂的相互作用强度,忽视了极化效应的重要性。基于这一认识,我们建议将 Drude 极化模型整合到主流力场中,并在碳酸盐、醚和腈电解质中验证其可行性。实验结果表明,这种方法大大提高了 CMD 模拟溶解结构的准确性。这项工作有望为电解质设计提供更可靠的 CMD 方法,使研究人员避免错误模拟结果的陷阱。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of Energy Chemistry
CHEMISTRY, APPLIED-CHEMISTRY, PHYSICAL
CiteScore
19.10
自引率
8.40%
发文量
3631
审稿时长
15 days
期刊介绍:
The Journal of Energy Chemistry, the official publication of Science Press and the Dalian Institute of Chemical Physics, Chinese Academy of Sciences, serves as a platform for reporting creative research and innovative applications in energy chemistry. It mainly reports on creative researches and innovative applications of chemical conversions of fossil energy, carbon dioxide, electrochemical energy and hydrogen energy, as well as the conversions of biomass and solar energy related with chemical issues to promote academic exchanges in the field of energy chemistry and to accelerate the exploration, research and development of energy science and technologies.
This journal focuses on original research papers covering various topics within energy chemistry worldwide, including:
Optimized utilization of fossil energy
Hydrogen energy
Conversion and storage of electrochemical energy
Capture, storage, and chemical conversion of carbon dioxide
Materials and nanotechnologies for energy conversion and storage
Chemistry in biomass conversion
Chemistry in the utilization of solar energy
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


