Nan Li, Zunkai Hu, Ning Zhang, Yining Liang, Yating Feng, Wanfu Ding, Lixin Cheng, Yuyan Zheng
{"title":"Pairwise analysis of gene expression for oral squamous cell carcinoma via a large-scale transcriptome integration","authors":"Nan Li, Zunkai Hu, Ning Zhang, Yining Liang, Yating Feng, Wanfu Ding, Lixin Cheng, Yuyan Zheng","doi":"10.1111/jcmm.70153","DOIUrl":null,"url":null,"abstract":"<p>Among all cancers occurring in the head and neck region, oral squamous cell carcinoma (OSCC) is the most common oral malignant tumours characterized by its aggressiveness and metastasis. The development of transcriptomics technology has greatly facilitated the diagnosis of various cancers. However, identifying genetic biomarkers is limited by data from a single batch of OSCC samples, and integrating analysis across different platforms remains a great challenge. In this study, we integrated five OSCC transcriptome datasets using an innovative strategy capable of mitigating batch effect, and extracting information from different datasets based on changes in the relative expression of gene pairs. By leveraging a machine learning method, we developed a prediction model including 27 differential gene pairs (DGPs) to discriminate OSCC from control samples, achieving an area under the receiver operating characteristic curve (AUC) of 0.8987 for the training set. Moreover, the model demonstrated commendable performance in four external validation sets, with AUCs of 0.9926, 0.9688, 0.8052 and 0.8565, respectively. Subsequently, a prognostic model was constructed based on six key gene pairs through univariate and multivariate Cox regression analysis. The AUCs of the model at 1-year and 3-year overall survival time prediction were 0.717 and 0.779 in an independent dataset. Our result demonstrates the effectiveness of this new method of integrating data and identifying DGPs. Using DGPs can significantly improve the performance of both diagnostic and prognostic models.</p>","PeriodicalId":101321,"journal":{"name":"JOURNAL OF CELLULAR AND MOLECULAR MEDICINE","volume":"28 20","pages":""},"PeriodicalIF":5.3000,"publicationDate":"2024-10-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/jcmm.70153","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"JOURNAL OF CELLULAR AND MOLECULAR MEDICINE","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/jcmm.70153","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
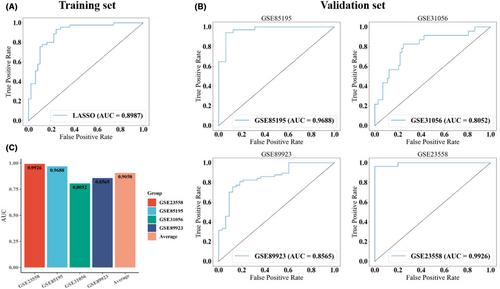
Among all cancers occurring in the head and neck region, oral squamous cell carcinoma (OSCC) is the most common oral malignant tumours characterized by its aggressiveness and metastasis. The development of transcriptomics technology has greatly facilitated the diagnosis of various cancers. However, identifying genetic biomarkers is limited by data from a single batch of OSCC samples, and integrating analysis across different platforms remains a great challenge. In this study, we integrated five OSCC transcriptome datasets using an innovative strategy capable of mitigating batch effect, and extracting information from different datasets based on changes in the relative expression of gene pairs. By leveraging a machine learning method, we developed a prediction model including 27 differential gene pairs (DGPs) to discriminate OSCC from control samples, achieving an area under the receiver operating characteristic curve (AUC) of 0.8987 for the training set. Moreover, the model demonstrated commendable performance in four external validation sets, with AUCs of 0.9926, 0.9688, 0.8052 and 0.8565, respectively. Subsequently, a prognostic model was constructed based on six key gene pairs through univariate and multivariate Cox regression analysis. The AUCs of the model at 1-year and 3-year overall survival time prediction were 0.717 and 0.779 in an independent dataset. Our result demonstrates the effectiveness of this new method of integrating data and identifying DGPs. Using DGPs can significantly improve the performance of both diagnostic and prognostic models.
期刊介绍:
The Journal of Cellular and Molecular Medicine serves as a bridge between physiology and cellular medicine, as well as molecular biology and molecular therapeutics. With a 20-year history, the journal adopts an interdisciplinary approach to showcase innovative discoveries.
It publishes research aimed at advancing the collective understanding of the cellular and molecular mechanisms underlying diseases. The journal emphasizes translational studies that translate this knowledge into therapeutic strategies. Being fully open access, the journal is accessible to all readers.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


