Alessandro Landi, Gaetano Ricci, Yoann Olivier, Amedeo Capobianco, Andrea Peluso
{"title":"Toward Efficient Modeling of Nonradiative Decay in Extended INVEST: Overcoming Computational Challenges in Quantum Dynamics Simulations.","authors":"Alessandro Landi, Gaetano Ricci, Yoann Olivier, Amedeo Capobianco, Andrea Peluso","doi":"10.1021/acs.jpclett.4c02713","DOIUrl":null,"url":null,"abstract":"<p><p>In recent years, an increasing number of fully organic molecules capable of thermally activated delayed fluorescence (TADF) have been reported, often with very small or even inverted singlet-triplet (INVEST) energy gaps. These molecules typically exhibit complex photophysics due to the close energy levels of multiple singlet and triplet states, which create various transition pathways toward emission. A predictive model for the rates of these transitions is thus essential for assessing the suitability of new materials for light-emitting devices. Quantum Dynamics (QD) calculations are ideal for this purpose, as they include quantum effects, without the limitations of first-order perturbative approaches, also allowing taking into account more than two electronic states at once. However, the huge computational demands of QD methodologies, especially for large molecules, currently limit their use as a standard tool. To address this problem, we here employ a strategy that allows us to include almost the whole set of the vibrational coordinates by selecting the key elements of the Hilbert space that significantly impact dynamics, thereby hugely reducing the computational burden. Application of this protocol to two relatively large INVEST molecules reveals that internal conversion in these systems is very fast, making indirect emissive pathways a possible channel for the population of the S<sub>1</sub> state. More importantly, this study demonstrates that the dynamics can be accurately described even with a significantly reduced vibrational space, thus allowing quantum dynamics calculations that yield accurate transition rates in a few minutes of computational time.</p>","PeriodicalId":62,"journal":{"name":"The Journal of Physical Chemistry Letters","volume":" ","pages":"11042-11050"},"PeriodicalIF":4.6000,"publicationDate":"2024-11-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry Letters","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpclett.4c02713","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/29 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
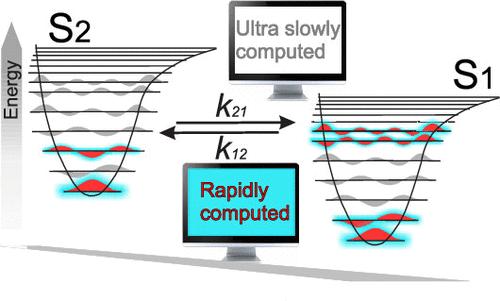
In recent years, an increasing number of fully organic molecules capable of thermally activated delayed fluorescence (TADF) have been reported, often with very small or even inverted singlet-triplet (INVEST) energy gaps. These molecules typically exhibit complex photophysics due to the close energy levels of multiple singlet and triplet states, which create various transition pathways toward emission. A predictive model for the rates of these transitions is thus essential for assessing the suitability of new materials for light-emitting devices. Quantum Dynamics (QD) calculations are ideal for this purpose, as they include quantum effects, without the limitations of first-order perturbative approaches, also allowing taking into account more than two electronic states at once. However, the huge computational demands of QD methodologies, especially for large molecules, currently limit their use as a standard tool. To address this problem, we here employ a strategy that allows us to include almost the whole set of the vibrational coordinates by selecting the key elements of the Hilbert space that significantly impact dynamics, thereby hugely reducing the computational burden. Application of this protocol to two relatively large INVEST molecules reveals that internal conversion in these systems is very fast, making indirect emissive pathways a possible channel for the population of the S1 state. More importantly, this study demonstrates that the dynamics can be accurately described even with a significantly reduced vibrational space, thus allowing quantum dynamics calculations that yield accurate transition rates in a few minutes of computational time.
期刊介绍:
The Journal of Physical Chemistry (JPC) Letters is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, chemical physicists, physicists, material scientists, and engineers. An important criterion for acceptance is that the paper reports a significant scientific advance and/or physical insight such that rapid publication is essential. Two issues of JPC Letters are published each month.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


