Comprehensive analysis of Mycobacterium tuberculosis genomes reveals genetic variations in bacterial virulence
IF 20.6
1区 医学
Q1 MICROBIOLOGY
引用次数: 0
Abstract
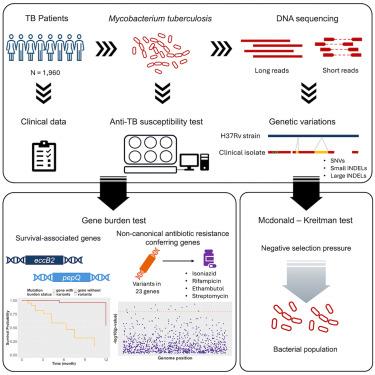
Tuberculosis, a disease caused by Mycobacterium tuberculosis (Mtb), is a significant health problem worldwide. Here, we developed a method to detect large insertions and deletions (indels), which have been generally understudied. Leveraging this framework, we performed a comprehensive analysis of single nucleotide variants and small and large indels across 1,960 Mtb clinical isolates. Comparing the distribution of variants demonstrated that gene disruptive variants are underrepresented in genes essential for bacterial survival. An evolutionary analysis revealed that Mtb genomes are enriched in partially deleterious mutations. Genome-wide association studies identified small and large deletions in eccB2 significantly associated with patient prognosis. Additionally, we unveil significant associations with antibiotic resistance in 23 non-canonical genes. Among these, large indels are primarily found in genetic regions of Rv1216c, Rv1217c, fadD11, and ctpD. This study provides a comprehensive catalog of genetic variations and highlights their potential impact for the future treatment and risk prediction of tuberculosis.
结核分枝杆菌基因组综合分析揭示了细菌毒力的遗传变异
结核病是由结核分枝杆菌(Mtb)引起的一种疾病,是世界性的重大健康问题。在这里,我们开发了一种检测大插入和大缺失(indels)的方法,但对这些问题的研究普遍不足。利用这一框架,我们对 1,960 株 Mtb 临床分离株中的单核苷酸变异和大小吲哚进行了全面分析。比较变异的分布情况表明,基因破坏性变异在细菌生存所必需的基因中所占比例较低。进化分析表明,Mtb基因组富含部分有害变异。全基因组关联研究发现,eccB2的小缺失和大缺失与患者的预后密切相关。此外,我们还揭示了 23 个非经典基因与抗生素耐药性的重要关联。其中,大缺失主要出现在 Rv1216c、Rv1217c、fadD11 和 ctpD 的基因区域。这项研究提供了一个全面的基因变异目录,并强调了它们对结核病未来治疗和风险预测的潜在影响。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Cell host & microbe
生物-微生物学
CiteScore
45.10
自引率
1.70%
发文量
201
审稿时长
4-8 weeks
期刊介绍:
Cell Host & Microbe is a scientific journal that was launched in March 2007. The journal aims to provide a platform for scientists to exchange ideas and concepts related to the study of microbes and their interaction with host organisms at a molecular, cellular, and immune level. It publishes novel findings on a wide range of microorganisms including bacteria, fungi, parasites, and viruses. The journal focuses on the interface between the microbe and its host, whether the host is a vertebrate, invertebrate, or plant, and whether the microbe is pathogenic, non-pathogenic, or commensal. The integrated study of microbes and their interactions with each other, their host, and the cellular environment they inhabit is a unifying theme of the journal. The published work in Cell Host & Microbe is expected to be of exceptional significance within its field and also of interest to researchers in other areas. In addition to primary research articles, the journal features expert analysis, commentary, and reviews on current topics of interest in the field.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


