Accurate protein-ligand binding free energy estimation using QM/MM on multi-conformers predicted from classical mining minima
IF 5.9
2区 化学
Q1 CHEMISTRY, MULTIDISCIPLINARY
引用次数: 0
Abstract
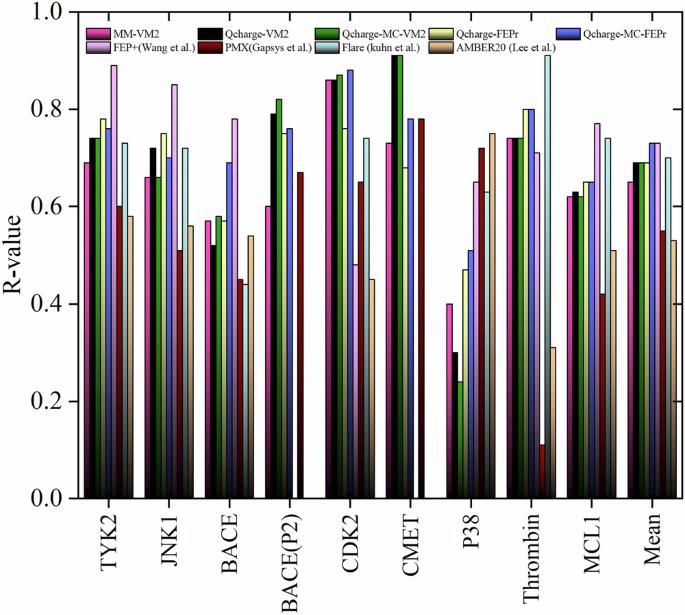
Accurate prediction of binding free energy is crucial for the rational design of drug candidates and understanding protein-ligand interactions. To address this, we have developed four protocols that combine QM/MM calculations and the mining minima (M2) method, tested on 9 targets and 203 ligands. Our protocols carry out free energy processing with or without conformational search on the selected conformers obtained from M2 calculations, where their force field atomic charge parameters are substituted with those obtained from a QM/MM calculation. The method achieved a high Pearson’s correlation coefficient (0.81) with experimental binding free energies across diverse targets, demonstrating its generality. Using a differential evolution algorithm with a universal scaling factor of 0.2, we achieved a low mean absolute error of 0.60 kcal mol-1. This performance surpasses many existing methods and is comparable to popular relative binding free energy techniques but at significantly lower computational cost. Binding free energy calculations are crucial in computational drug discovery, however, the current alchemical free energy perturbation (FEP) requires large computational capabilities to achieve high accuracy. Here, the authors develop an alternative method by combining QM/MM and the mining minima method to predict free energies at lower computational cost and with comparable accuracy to FEP-based methods.
利用 QM/MM 对经典采矿极小值预测的多构型进行精确的蛋白质配体结合自由能估算
准确预测结合自由能对于合理设计候选药物和理解蛋白质配体相互作用至关重要。为此,我们开发了四种结合 QM/MM 计算和挖掘最小值(M2)方法的方案,并在 9 个目标和 203 种配体上进行了测试。我们的方案对通过 M2 计算获得的选定构象进行了自由能处理(有构象搜索或无构象搜索),将其力场原子电荷参数替换为通过 QM/MM 计算获得的参数。该方法与不同目标的实验结合自由能之间的皮尔逊相关系数高达 0.81,证明了其通用性。使用通用缩放因子为 0.2 的微分进化算法,我们实现了 0.60 kcal mol-1 的低平均绝对误差。这一性能超越了许多现有方法,可与流行的相对结合自由能技术相媲美,但计算成本却大大降低。结合自由能计算在计算药物发现中至关重要,然而,目前的炼金术自由能扰动(FEP)需要庞大的计算能力才能达到高精度。在此,作者开发了一种替代方法,将 QM/MM 与采矿极小值法相结合,以较低的计算成本预测自由能,其精确度与基于 FEP 的方法相当。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊
Communications Chemistry
Chemistry-General Chemistry
CiteScore
7.70
自引率
1.70%
发文量
146
审稿时长
13 weeks
期刊介绍:
Communications Chemistry is an open access journal from Nature Research publishing high-quality research, reviews and commentary in all areas of the chemical sciences. Research papers published by the journal represent significant advances bringing new chemical insight to a specialized area of research. We also aim to provide a community forum for issues of importance to all chemists, regardless of sub-discipline.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


