Theoretical mechanistic insights on the thermal and acid-catalyzed rearrangements of N-methyl-N-nitroanilines†
IF 2.9
3区 化学
Q1 CHEMISTRY, ORGANIC
引用次数: 0
Abstract
The thermal and acid-catalyzed rearrangement mechanisms of N-methyl-N-nitroanilines were theoretically investigated via density functional theory (DFT) calculations for all possible proposed mechanisms. The results indicate that the thermal rearrangement of N-methyl-N-nitroanilines undergoes a radical pair complex mechanism through the homolysis of their N–N bond to generate a radical pair complex and the recombination of the radical pairs followed by aromatization. For the acid-catalyzed rearrangements, N-methyl-N-nitroanilines are first protonated on the nitrogen atom of their aniline moiety and then generate protonated N-methyl-O-nitroso-N-phenylhydroxylamines through a three-membered spirocyclic oxadiaziridine transition state. The N-protonated N-methyl-O-nitroso-N-phenylhydroxylamines favor homolytic dissociation to generate N-methylaniline cationic radical and nitrogen dioxide complexes, which further combine together and aromatize to afford protonated N-methyl-o-nitroanilines and N-methyl-p-nitroanilines, respectively. The radical pair complexes are more stable than the corresponding solvent-caged radical pairs. The thermal rearrangements require higher activation energy than the corresponding acid-catalyzed rearrangements.
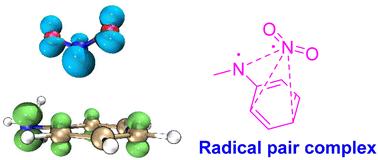
关于 N-甲基-N-硝基苯胺热重排和酸催化重排的理论机理见解。
通过密度泛函理论(DFT)计算,对所有可能提出的 N-甲基-N-硝基苯胺的热重排和酸催化重排机理进行了理论研究。结果表明,N-甲基-N-硝基苯胺的热重排经历了一个自由基对复合物机制,通过 N-N 键的均裂生成自由基对复合物,自由基对重组后发生芳香化。在酸催化重排中,N-甲基-N-硝基苯胺首先在其苯胺分子的氮原子上质子化,然后通过三元螺环噁二嗪过渡态生成质子化的 N-甲基-O-亚硝基-N-苯基羟胺。N-质子化的 N-甲基-O-亚硝基-N-苯基羟胺有利于同解,生成 N-甲基苯胺阳离子自由基和二氧化氮络合物,它们进一步结合在一起并芳香化,分别生成质子化的 N-甲基-邻硝基苯胺和 N-甲基-对硝基苯胺。这些自由基对复合物比相应的溶剂笼自由基对更加稳定。热重排所需的活化能高于相应的酸催化重排。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Organic & Biomolecular Chemistry
化学-有机化学
CiteScore
5.50
自引率
9.40%
发文量
1056
审稿时长
1.3 months
期刊介绍:
Organic & Biomolecular Chemistry is an international journal using integrated research in chemistry-organic chemistry. Founded in 2003 by the Royal Society of Chemistry, the journal is published in Semimonthly issues and has been indexed by SCIE, a leading international database. The journal focuses on the key research and cutting-edge progress in the field of chemistry-organic chemistry, publishes and reports the research results in this field in a timely manner, and is committed to becoming a window and platform for rapid academic exchanges among peers in this field. The journal's impact factor in 2023 is 2.9, and its CiteScore is 5.5.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


