Accurate Calculation of Interatomic Forces with Neural Networks Based on a Generative Transformer Architecture
IF 5.5
1区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
Using neural networks to express electronic wave functions represents a new paradigm for solving the Schrödinger equation in quantum chemistry. For practical quantum chemistry simulations, one needs to know not only energies of molecules, but also accurate forces acting on constituent atoms. In this work, we achieve the accurate calculation of interatomic forces on QiankunNet, a platform that combines transformer-based deep neural networks with efficient batched autoregressive sampling. Our approach permits the application of the Hellmann–Feynman theorem to force calculations without introducing corrective Pulay terms. The results show that the calculated interatomic forces are in close agreement with those derived from the full configuration interaction method, irrespective of whether the system is a simple molecule or a strongly correlated electron system like a linear hydrogen chain. Furthermore, the calculated interatomic forces are employed for atomic relaxation in the torsional rotation process of ethylene, and the energy barrier obtained from the scanned potential energy surface is in excellent agreement with the experiment. Our work contributes to the application of artificial intelligence to broader quantum chemistry simulations, such as modeling challenging chemical transformations where electron correlations are difficult to describe.
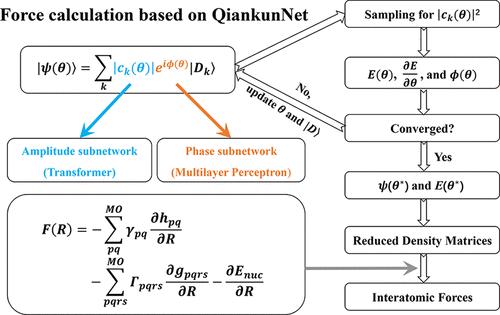
利用基于生成变压器架构的神经网络精确计算原子间作用力
使用神经网络来表达电子波函数是解决量子化学中薛定谔方程的一种新模式。在实际的量子化学模拟中,人们不仅需要知道分子的能量,还需要知道作用在组成原子上的精确力。在这项工作中,我们在乾坤网上实现了原子间作用力的精确计算。乾坤网是一个将基于变压器的深度神经网络与高效批次自回归采样相结合的平台。我们的方法允许将赫尔曼-费曼定理应用于力计算,而无需引入校正普雷项。结果表明,无论系统是简单分子还是线性氢链等强相关电子系统,计算出的原子间作用力都与全构型相互作用方法得出的原子间作用力非常接近。此外,在乙烯的扭转旋转过程中,利用计算出的原子间力进行原子弛豫,从扫描势能面得到的能量势垒与实验结果非常吻合。我们的工作有助于将人工智能应用于更广泛的量子化学模拟,例如模拟电子关联难以描述的高难度化学转化。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of Chemical Theory and Computation
化学-物理:原子、分子和化学物理
CiteScore
9.90
自引率
16.40%
发文量
568
审稿时长
1 months
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


