Cooperative Transport of Lithium in Disordered Li10MP2S12 (M = Sn, Si) Electrolytes for Li-Ion Batteries
IF 7.2
2区 材料科学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
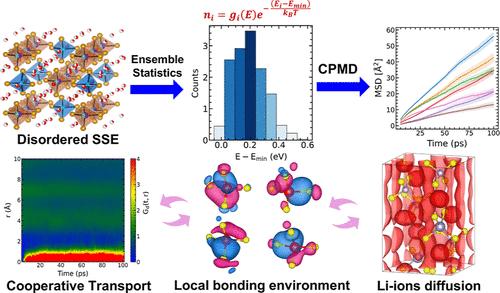
Disorder in sulfide solid-state electrolytes significantly impacts chemical bonding, affecting electrochemical properties and interface stability. Li10GeP2S12, a prominent sulfide electrolyte, is expensive and has limited interfacial stability, so substituting Ge with earth-abundant elements, such as Sn and Si, could be more practical. However, a thorough understanding of the kinetics and chemical bonding nature of Li in the Sn/Si-substituted systems is missing owing to the complexity associated with disordered sublattice in these materials. We use isothermal–isobaric ensemble Car–Parrinello molecular dynamics to evaluate configuration-dependent tracer and charged diffusivities and activation energies for lithium-ion migration in disordered configurations of Li10SiP2S12 (LSiPS) and Li10SnP2S12 (LSnPS) obtained using ensemble statistics. The study uses Li-ion probability density and maximally localized Wannier orbital analysis to determine how temperature and Sn and Si cations affect Li-ion migration. Our findings indicate that higher temperatures enhance Li-ion mobility by enabling more diffusion pathways. The disordered LSiPS and LSnPS electronic structure shows a Kohn–Sham band gap of 2.4 eV for LSiPS and 2 eV for LSnPS, of the most probable configuration across 500 configurations, suggesting a wider electrolyte window for LSiPS. Additionally, Wannier function visualizations demonstrated the significant impact of locality and temperature on the dynamic nature of bonding states of migrating Li ions.
用于锂离子电池的无序 Li10MP2S12(M = Sn,Si)电解质中锂的协同传输
硫化物固态电解质中的无序状态会严重影响化学键,从而影响电化学特性和界面稳定性。Li10GeP2S12 是一种重要的硫化物电解质,价格昂贵且界面稳定性有限,因此用锡和硅等地球富集元素替代 Ge 可能更加实用。然而,由于这些材料中无序亚晶格的复杂性,人们对 Sn/Si 取代体系中 Li 的动力学和化学键性质还缺乏透彻的了解。我们使用等温等压集合 Car-Parrinello 分子动力学来评估构型相关的示踪剂和带电扩散量,以及利用集合统计获得的 Li10SiP2S12 (LSiPS) 和 Li10SnP2S12 (LSnPS) 无序构型中锂离子迁移的活化能。该研究利用锂离子概率密度和最大局部万尼尔轨道分析来确定温度以及锡和硅阳离子如何影响锂离子迁移。我们的研究结果表明,较高的温度能提供更多的扩散途径,从而提高锂离子迁移率。无序 LSiPS 和 LSnPS 电子结构显示,LSiPS 的 Kohn-Sham 带隙为 2.4 eV,LSnPS 为 2 eV。此外,万尼尔函数可视化显示了位置和温度对迁移锂离子成键状态动态性质的重大影响。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Chemistry of Materials
工程技术-材料科学:综合
CiteScore
14.10
自引率
5.80%
发文量
929
审稿时长
1.5 months
期刊介绍:
The journal Chemistry of Materials focuses on publishing original research at the intersection of materials science and chemistry. The studies published in the journal involve chemistry as a prominent component and explore topics such as the design, synthesis, characterization, processing, understanding, and application of functional or potentially functional materials. The journal covers various areas of interest, including inorganic and organic solid-state chemistry, nanomaterials, biomaterials, thin films and polymers, and composite/hybrid materials. The journal particularly seeks papers that highlight the creation or development of innovative materials with novel optical, electrical, magnetic, catalytic, or mechanical properties. It is essential that manuscripts on these topics have a primary focus on the chemistry of materials and represent a significant advancement compared to prior research. Before external reviews are sought, submitted manuscripts undergo a review process by a minimum of two editors to ensure their appropriateness for the journal and the presence of sufficient evidence of a significant advance that will be of broad interest to the materials chemistry community.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


