Yichuan Liu, Hui-Qi Qu, Xiao Chang, Frank D Mentch, Haijun Qiu, Kenny Nguyen, Kayleigh Ostberg, Tiancheng Wang, Joseph Glessner, Hakon Hakonarson
{"title":"Deciphering protective genomic factors of tumor development in pediatric Down syndrome via deep learning approach to whole genome and RNA sequencing","authors":"Yichuan Liu, Hui-Qi Qu, Xiao Chang, Frank D Mentch, Haijun Qiu, Kenny Nguyen, Kayleigh Ostberg, Tiancheng Wang, Joseph Glessner, Hakon Hakonarson","doi":"10.1002/cac2.12612","DOIUrl":null,"url":null,"abstract":"<p>Childhood solid tumors represent a significant public health challenge worldwide, with approximately 15,000 new cases annually in the United States and an estimated 300,000 globally. Down syndrome (DS), a genetic disorder characterized by an extra full or partial copy of chromosome 21, results in distinctive developmental and physical features. Notably, individuals with DS exhibit a remarkable resilience against solid tumors compared to the general population, with an overall standardized incidence ratio (SIR) of 0.45, despite their increased susceptibility to hematologic malignancies [<span>1</span>]. This paradoxical observation has spurred extensive research aimed at uncovering the biological underpinnings of this natural resistance to solid cancers. Current theories suggest that the overexpression of specific genes on chromosome 21 may confer protective benefits (e.g. <i>RCAN1</i> contributes to antiangiogenic effects), and alterations in immune system function may enhance apoptosis and DNA repair pathways in individuals with trisomy 21 DS [<span>2</span>]. The well-established epigenetic effects of trisomy 21, which influence the entire genome, are another potential contributor to the reduced risk of solid tumors [<span>3</span>]. Nonetheless, these hypotheses face significant challenges, such as the potential oversimplification of complex genetic interactions and the lack of comprehensive genome-wide analyses. This study seeks to critically evaluate the correlations between genomic variants and cancer clinical phenotypes in patients with DS, and proposes directions for future research into the genetic and molecular mechanisms that confer cancer resistance in DS, potentially transforming our understanding and treatment of pediatric cancers.</p><p>We conducted an innovative unbiased data-driven analysis in 2,452 whole-genome sequencing (WGS) samples with both DS individuals (<i>n</i> = 635) and pediatric oncology cases (<i>n</i> = 280) within the Gabriella Miller Kids First program project (https://kidsfirstdrc.org/) housed at the Children's Hospital of Philadelphia (Supplementary Figure S1). Additionally, 284 RNA sequencing samples from human peripheral blood mononuclear cells (PBMCs), a subset of WGS samples, were also analyzed, offering unprecedented insights into the complex interplay of genetic and immunological factors influencing cancer resistance.</p><p>The importance of each variant was calculated using deep learning algorithms, and their corresponding weights to DS cancer were generated based on linear algebra models as described in the Supplementary Materials and Methods. There were 2,523 unique cancer protective variants identified based on deep learning algorithms combined with linear algebra models in exonic, intronic, non-coding RNA and 5’untranslated region (5’UTR) regions. The prevalence for cancer protective variants in the DS cancer group (89.2%) is significantly higher compared to non-DS cancer individuals (58.1%) (<i>P</i> = 1.11 × 10<sup>−40</sup>), indicating that DS individuals may be protected against solid tumors by cancer protective variants identified in this study. The functional enrichment analysis revealed cancer development-related pathways for distinct categories of variants identified by WGS (Supplementary Figure S2). Of note, the functional terms differed between protective and predisposing variants, irrespective of the databases used, including Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), Protein Analysis Through Evolutionary Relationships (PANTHER), database of reactions, pathways and biological processes (Reactome), and wiki-based resource for collection, maintenance and distribution of biological pathways (WikiPath). A total of 121 genes exhibited overlapping between the cancer-protective and cancer-predisposing variants and their corresponding genes (Supplementary Table S1-S2). This intersection of genes, with a very low likelihood of occurring by chance (<i>P =</i> 0.002), attained statistical significance within cancer essential pathways such as the p53 pathway (False discovery rate [FDR] <i><</i> 0.001) (Figure 1A). These outcomes aligned with expectations regarding the multifunctional roles of genes in the intricate processes of tumor development [<span>4</span>]. Variant type distributions revealed distinctive patterns within the 121 genes that are common to both categories (Figure 1B). Specifically, cancer-predisposing variants were more prevalent in exon regions for nonsynonymous or synonymous variants, in contrast, cancer-protective variants exhibited a higher prevalence in non-coding regions, suggesting regulatory roles in tumor development. Additionally, cancer-protective variants demonstrated earlier transcription activity than their predisposing counterparts (Figure 1C). These findings suggested that overlapping genes may serve dual roles as either cancer enhancers or suppressors, depending on the functional effects of the minor allele of a genetic variant. The nature of their impact is contingent upon various factors, including but not limited to variant categories and the genomic loci where these variants reside. To leverage prior cancer genetic knowledge, we integrated data from the Catalogue of Somatic Mutations in Cancer (COSMIC) and pediatric cancer driver genes to construct a curated list comprising 830 known cancer genes. Among the 121 overlapping genes, 18 were identified within this established cancer driver gene set. For instance, the isocitrate dehydrogenase 1 (<i>IDH1</i>) gene, associated with frequent mutations across various cancer types and tissues of origin [<span>5</span>], harbors a cancer-protective variant in the 5’UTR region (chr2:208254188-G-A) and a cancer-predisposing variant in the tail region (intron 8 of 9, chr2:208239246-A-G) (Figure 1D). Another example is gene inhibitor of DNA binding 3 (<i>ID3</i>), a member of the ID protein family implicated in cancer development, stemness, and metastasis [<span>6</span>], manifesting a protective variant in the 5’UTR (chr1:23559494-T-C) and a predisposing variant in the tail of exon 1 as a nonsynonymous mutation (chr1:23559171-C-G) (Figure 1D). Conversely, <i>UBQLN1</i>, a gene with cancer-protective variants located in the gene's tail (last intron) (Figure 1D), suppresses cancer stem cell-like traits in non-small cell lung cancer cells by regulating reactive oxygen species homeostasis [<span>7</span>]. For 148 cancer predisposing variants identified in cancer driver genes, the prevalence is 25% in DS cancer patients and 41.3% in non-DS cancer patients, suggested that DS population are under protections for cancer, and DS cancer patients may have different tumor development mechanisms compared to non-DS children.</p><p>Analysis of the 284 RNA-sequencing (RNA-seq) PBMC samples yielded a highly valuable insight into the direction and magnitude of gene expression corresponding to selected variants. The categorization of cancer-protective and cancer-predisposing gene sets was further stratified into four subgroups for functional enrichment analysis based on direction. Notably, genes possessing cancer-protective variants that were down-regulated (suppressed) in cancer patients exhibited much stronger signals compared to other sets (Figure 1E-F). This phenomenon, especially evident in pathways relevant to tumor development such as Proteoglycans in cancer (FDR <i><</i> 0.001) and Central carbon metabolism in cancer (FDR <i><</i> 0.001), emphasized the significance of protective variants as dominant factors in tumor development among DS patients. A total of 1,785 genes with cancer-protective variants were presented, and genes with highest fold-changes in RNA-seq results while known as cancer driver genes were shown in Supplementary Table S3. Among 1,785 genes, 983 genes were found to be downregulated in cancer patients. Intriguingly, 86 of these down-regulated genes, mapped by 109 cancer-protective variants (Supplementary Table S4-S5), were identified as known cancer driver genes. These 86 suppressed genes in cancer patients with protective variants were not only associated with essential cancer pathways (FDR <i><</i> 0.001) (Supplementary Figure S3A) but are also significantly enriched in cancer treatment response pathways, including epidermal growth factor receptor (<i>EGFR</i>) tyrosine kinase inhibitor resistance (FDR <i><</i> 0.001) and programmed death-ligand 1 (PD-L1) expression and programmed cell death protein 1 (PD-1) checkpoint pathway in cancer (FDR <i><</i> 0.001) (Supplementary Figure S3B-C). Referring to the National Cancer Institute's approved drug list, gene targets within the 86 genes are shown in Supplementary Table S6.</p><p>Our study suggests that genes with cancer-protective variants down-regulated in cancer patients may act as a critical factor for the protective mechanism against solid tumors in DS patients. A nonsynonymous cancer-protective variant (chr7:55205451-A-C) in <i>EGFR</i> exon 28, resulting in the truncation of the C-terminal domain of <i>EGFR</i>, has been previously reported to be associated with glioblastoma multiforme (GBM) patients [<span>8</span>]. This variant in <i>EGFR</i> demonstrated a high correlation coefficient (correlation coefficient > 0.7) with another nonsynonymous cancer-protective variant (chr7:55205451-A-C) in exon 8 of <i>SEPTIN14</i>, and the <i>EGFR</i>-<i>SEPTIN14</i> fusion has been linked to glioblastoma with Icotinib-sensitive drug responses [<span>9</span>]. Furthermore, a variant (chr7:55255565-T-C) for the ncRNA <i>ELDR</i> (<i>EGFR</i> long non-coding downstream RNA) was identified in exon 1, with a previous study showing that knockdown of <i>ELDR</i> resulted in the downregulation of <i>EGFR</i>, leading to the inactivation of downstream molecules, and it is considered a therapeutic potential target in cancer [<span>10</span>]. For the remaining genes down-regulated in cancer patients with cancer-protective variants that are not recognized as cancer driver genes, they also exhibit enrichment in cancer-related pathways, including the AMPK signaling pathway (FDR <i>=</i> 0.032), PI3K-Akt signaling pathway (FDR <i>=</i> 0.004), and Focal adhesion (FDR <i>=</i> 0.044) (Supplementary Figure S3D-F).</p><p>This study significantly advances our understanding of how genetic factors associated with DS contribute to a reduced risk of solid tumor development. Through an examination of more than 2,000 WGS samples, we identified genetic variants playing important roles in either protecting or predisposing individuals to cancer. With the revealed correlations between protective variants, cancer mechanisms, and treatment response pathways, our findings warrant exploring new therapeutic interventions at the gene or pathway level. The development of targeted therapies, inspired by the natural protective mechanisms found in DS individuals, could transform the landscape of cancer treatment, with far-reaching implications extending beyond the DS population.</p><p><i>Conceptualization and supervision</i>: Yichuan Liu and Hakon Hakonarson. <i>Literature search</i>: Yichuan Liu. <i>Data preparation and analysis</i>: Yichuan Liu, Hui-Qi Qu, Xiao Chang, Frank D Mentch, Haijun Qiu, Kenny Nguyen, Kayleigh Ostberg, and Tiancheng Wang. <i>Data interpretation</i>: Yichuan Liu, Hui-Qi Qu, Xiao Chang, Joseph Glessner, and Hakon Hakonarson. <i>Original draft writing</i>: Yichuan Liu. <i>Review and revision</i>: Yichuan Liu, Hui-Qi Qu, and Hakon Hakonarson. All authors read and approved the final manuscript.</p><p>The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.</p><p>The study was supported by the Institutional Development Funds from the Children's Hospital of Philadelphia to the Center for Applied Genomics, and The Children's Hospital of Philadelphia Endowed Chair in Genomic Research to Hakon Hakonarson.</p><p>We confirm that all methods were carried out in accordance with relevant guidelines and regulations. All experimental protocols were approved by the Institutional Review Board (IRB) of the Children's Hospital of Philadelphia (CHOP) with the IRB number: IRB 16-013278.</p><p>Informed consent was obtained from all subjects. If subjects are under 18, consent was obtained from a parent and/or legal guardian with assent from the child if 7 years or older.</p>","PeriodicalId":9495,"journal":{"name":"Cancer Communications","volume":"44 11","pages":"1374-1378"},"PeriodicalIF":24.9000,"publicationDate":"2024-10-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/cac2.12612","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cancer Communications","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/cac2.12612","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"ONCOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Childhood solid tumors represent a significant public health challenge worldwide, with approximately 15,000 new cases annually in the United States and an estimated 300,000 globally. Down syndrome (DS), a genetic disorder characterized by an extra full or partial copy of chromosome 21, results in distinctive developmental and physical features. Notably, individuals with DS exhibit a remarkable resilience against solid tumors compared to the general population, with an overall standardized incidence ratio (SIR) of 0.45, despite their increased susceptibility to hematologic malignancies [1]. This paradoxical observation has spurred extensive research aimed at uncovering the biological underpinnings of this natural resistance to solid cancers. Current theories suggest that the overexpression of specific genes on chromosome 21 may confer protective benefits (e.g. RCAN1 contributes to antiangiogenic effects), and alterations in immune system function may enhance apoptosis and DNA repair pathways in individuals with trisomy 21 DS [2]. The well-established epigenetic effects of trisomy 21, which influence the entire genome, are another potential contributor to the reduced risk of solid tumors [3]. Nonetheless, these hypotheses face significant challenges, such as the potential oversimplification of complex genetic interactions and the lack of comprehensive genome-wide analyses. This study seeks to critically evaluate the correlations between genomic variants and cancer clinical phenotypes in patients with DS, and proposes directions for future research into the genetic and molecular mechanisms that confer cancer resistance in DS, potentially transforming our understanding and treatment of pediatric cancers.
We conducted an innovative unbiased data-driven analysis in 2,452 whole-genome sequencing (WGS) samples with both DS individuals (n = 635) and pediatric oncology cases (n = 280) within the Gabriella Miller Kids First program project (https://kidsfirstdrc.org/) housed at the Children's Hospital of Philadelphia (Supplementary Figure S1). Additionally, 284 RNA sequencing samples from human peripheral blood mononuclear cells (PBMCs), a subset of WGS samples, were also analyzed, offering unprecedented insights into the complex interplay of genetic and immunological factors influencing cancer resistance.
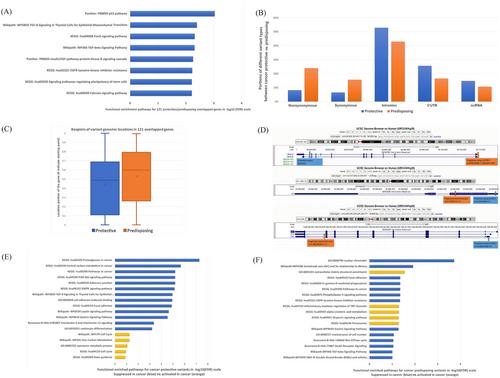
The importance of each variant was calculated using deep learning algorithms, and their corresponding weights to DS cancer were generated based on linear algebra models as described in the Supplementary Materials and Methods. There were 2,523 unique cancer protective variants identified based on deep learning algorithms combined with linear algebra models in exonic, intronic, non-coding RNA and 5’untranslated region (5’UTR) regions. The prevalence for cancer protective variants in the DS cancer group (89.2%) is significantly higher compared to non-DS cancer individuals (58.1%) (P = 1.11 × 10−40), indicating that DS individuals may be protected against solid tumors by cancer protective variants identified in this study. The functional enrichment analysis revealed cancer development-related pathways for distinct categories of variants identified by WGS (Supplementary Figure S2). Of note, the functional terms differed between protective and predisposing variants, irrespective of the databases used, including Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), Protein Analysis Through Evolutionary Relationships (PANTHER), database of reactions, pathways and biological processes (Reactome), and wiki-based resource for collection, maintenance and distribution of biological pathways (WikiPath). A total of 121 genes exhibited overlapping between the cancer-protective and cancer-predisposing variants and their corresponding genes (Supplementary Table S1-S2). This intersection of genes, with a very low likelihood of occurring by chance (P = 0.002), attained statistical significance within cancer essential pathways such as the p53 pathway (False discovery rate [FDR] < 0.001) (Figure 1A). These outcomes aligned with expectations regarding the multifunctional roles of genes in the intricate processes of tumor development [4]. Variant type distributions revealed distinctive patterns within the 121 genes that are common to both categories (Figure 1B). Specifically, cancer-predisposing variants were more prevalent in exon regions for nonsynonymous or synonymous variants, in contrast, cancer-protective variants exhibited a higher prevalence in non-coding regions, suggesting regulatory roles in tumor development. Additionally, cancer-protective variants demonstrated earlier transcription activity than their predisposing counterparts (Figure 1C). These findings suggested that overlapping genes may serve dual roles as either cancer enhancers or suppressors, depending on the functional effects of the minor allele of a genetic variant. The nature of their impact is contingent upon various factors, including but not limited to variant categories and the genomic loci where these variants reside. To leverage prior cancer genetic knowledge, we integrated data from the Catalogue of Somatic Mutations in Cancer (COSMIC) and pediatric cancer driver genes to construct a curated list comprising 830 known cancer genes. Among the 121 overlapping genes, 18 were identified within this established cancer driver gene set. For instance, the isocitrate dehydrogenase 1 (IDH1) gene, associated with frequent mutations across various cancer types and tissues of origin [5], harbors a cancer-protective variant in the 5’UTR region (chr2:208254188-G-A) and a cancer-predisposing variant in the tail region (intron 8 of 9, chr2:208239246-A-G) (Figure 1D). Another example is gene inhibitor of DNA binding 3 (ID3), a member of the ID protein family implicated in cancer development, stemness, and metastasis [6], manifesting a protective variant in the 5’UTR (chr1:23559494-T-C) and a predisposing variant in the tail of exon 1 as a nonsynonymous mutation (chr1:23559171-C-G) (Figure 1D). Conversely, UBQLN1, a gene with cancer-protective variants located in the gene's tail (last intron) (Figure 1D), suppresses cancer stem cell-like traits in non-small cell lung cancer cells by regulating reactive oxygen species homeostasis [7]. For 148 cancer predisposing variants identified in cancer driver genes, the prevalence is 25% in DS cancer patients and 41.3% in non-DS cancer patients, suggested that DS population are under protections for cancer, and DS cancer patients may have different tumor development mechanisms compared to non-DS children.
Analysis of the 284 RNA-sequencing (RNA-seq) PBMC samples yielded a highly valuable insight into the direction and magnitude of gene expression corresponding to selected variants. The categorization of cancer-protective and cancer-predisposing gene sets was further stratified into four subgroups for functional enrichment analysis based on direction. Notably, genes possessing cancer-protective variants that were down-regulated (suppressed) in cancer patients exhibited much stronger signals compared to other sets (Figure 1E-F). This phenomenon, especially evident in pathways relevant to tumor development such as Proteoglycans in cancer (FDR < 0.001) and Central carbon metabolism in cancer (FDR < 0.001), emphasized the significance of protective variants as dominant factors in tumor development among DS patients. A total of 1,785 genes with cancer-protective variants were presented, and genes with highest fold-changes in RNA-seq results while known as cancer driver genes were shown in Supplementary Table S3. Among 1,785 genes, 983 genes were found to be downregulated in cancer patients. Intriguingly, 86 of these down-regulated genes, mapped by 109 cancer-protective variants (Supplementary Table S4-S5), were identified as known cancer driver genes. These 86 suppressed genes in cancer patients with protective variants were not only associated with essential cancer pathways (FDR < 0.001) (Supplementary Figure S3A) but are also significantly enriched in cancer treatment response pathways, including epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor resistance (FDR < 0.001) and programmed death-ligand 1 (PD-L1) expression and programmed cell death protein 1 (PD-1) checkpoint pathway in cancer (FDR < 0.001) (Supplementary Figure S3B-C). Referring to the National Cancer Institute's approved drug list, gene targets within the 86 genes are shown in Supplementary Table S6.
Our study suggests that genes with cancer-protective variants down-regulated in cancer patients may act as a critical factor for the protective mechanism against solid tumors in DS patients. A nonsynonymous cancer-protective variant (chr7:55205451-A-C) in EGFR exon 28, resulting in the truncation of the C-terminal domain of EGFR, has been previously reported to be associated with glioblastoma multiforme (GBM) patients [8]. This variant in EGFR demonstrated a high correlation coefficient (correlation coefficient > 0.7) with another nonsynonymous cancer-protective variant (chr7:55205451-A-C) in exon 8 of SEPTIN14, and the EGFR-SEPTIN14 fusion has been linked to glioblastoma with Icotinib-sensitive drug responses [9]. Furthermore, a variant (chr7:55255565-T-C) for the ncRNA ELDR (EGFR long non-coding downstream RNA) was identified in exon 1, with a previous study showing that knockdown of ELDR resulted in the downregulation of EGFR, leading to the inactivation of downstream molecules, and it is considered a therapeutic potential target in cancer [10]. For the remaining genes down-regulated in cancer patients with cancer-protective variants that are not recognized as cancer driver genes, they also exhibit enrichment in cancer-related pathways, including the AMPK signaling pathway (FDR = 0.032), PI3K-Akt signaling pathway (FDR = 0.004), and Focal adhesion (FDR = 0.044) (Supplementary Figure S3D-F).
This study significantly advances our understanding of how genetic factors associated with DS contribute to a reduced risk of solid tumor development. Through an examination of more than 2,000 WGS samples, we identified genetic variants playing important roles in either protecting or predisposing individuals to cancer. With the revealed correlations between protective variants, cancer mechanisms, and treatment response pathways, our findings warrant exploring new therapeutic interventions at the gene or pathway level. The development of targeted therapies, inspired by the natural protective mechanisms found in DS individuals, could transform the landscape of cancer treatment, with far-reaching implications extending beyond the DS population.
Conceptualization and supervision: Yichuan Liu and Hakon Hakonarson. Literature search: Yichuan Liu. Data preparation and analysis: Yichuan Liu, Hui-Qi Qu, Xiao Chang, Frank D Mentch, Haijun Qiu, Kenny Nguyen, Kayleigh Ostberg, and Tiancheng Wang. Data interpretation: Yichuan Liu, Hui-Qi Qu, Xiao Chang, Joseph Glessner, and Hakon Hakonarson. Original draft writing: Yichuan Liu. Review and revision: Yichuan Liu, Hui-Qi Qu, and Hakon Hakonarson. All authors read and approved the final manuscript.
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The study was supported by the Institutional Development Funds from the Children's Hospital of Philadelphia to the Center for Applied Genomics, and The Children's Hospital of Philadelphia Endowed Chair in Genomic Research to Hakon Hakonarson.
We confirm that all methods were carried out in accordance with relevant guidelines and regulations. All experimental protocols were approved by the Institutional Review Board (IRB) of the Children's Hospital of Philadelphia (CHOP) with the IRB number: IRB 16-013278.
Informed consent was obtained from all subjects. If subjects are under 18, consent was obtained from a parent and/or legal guardian with assent from the child if 7 years or older.
期刊介绍:
Cancer Communications is an open access, peer-reviewed online journal that encompasses basic, clinical, and translational cancer research. The journal welcomes submissions concerning clinical trials, epidemiology, molecular and cellular biology, and genetics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


