Discovery of quinazoline-benzothiazole derivatives as novel potent protease-activated receptor 4 antagonists with improved pharmacokinetics and low bleeding liability
IF 6
2区 医学
Q1 CHEMISTRY, MEDICINAL
引用次数: 0
Abstract
Protease-activated receptor 4 (PAR4) plays a critical role in the development of pathological thrombosis, and targeting PAR4 is considered a promising strategy for improving antiplatelet therapies. Here, we reported the design of a series of quinazoline-benzothiazole-based PAR4 antagonists using a scaffold-hopping strategy. Systematic structure-activity relationship exploration leads to the discovery of compounds 20f and 20g, which displayed optimal activity (h. PAR4-AP PRP IC50 = 6.39 nM and 3.45 nM, respectively) on human platelets and high selectivity for PAR4. Both of them also showed excellent metabolic stability in human liver microsomes (compound 20f, T1/2 = 249.83 min, compound 20g, T1/2 = 282.60 min) and favourable PK profiles in rats (compound 20f, T1/2 = 5.16 h, F = 50.5 %, compound 20g, T1/2 = 7.05 h, F = 27.3 %). More importantly, neither compound prolonged the bleeding time in the mouse tail-cutting model (10 mg/kg, p.o.). These results suggest that these compounds have great potential for use in antiplatelet therapies.
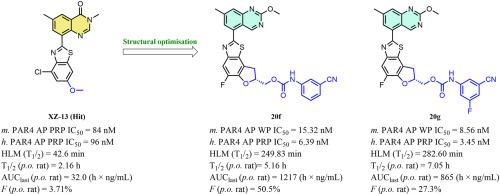
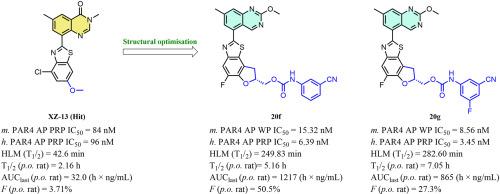
发现喹唑啉-苯并噻唑衍生物作为新型强效蛋白酶激活受体 4 拮抗剂,改善药代动力学,降低出血风险
蛋白酶活化受体 4(PAR4)在病理性血栓形成中起着关键作用,靶向 PAR4 被认为是改进抗血小板疗法的一种有前途的策略。在此,我们报告了利用支架跳转策略设计的一系列基于喹唑啉-苯并噻唑的 PAR4 拮抗剂。通过系统的结构-活性关系探索,我们发现了化合物 20f 和 20g,它们在人血小板上显示出最佳活性(h. PAR4-AP PRP IC50 = 6.39 nM 和 3.45 nM),并且对 PAR4 具有高选择性。这两种化合物在人肝脏微粒体中也表现出极佳的代谢稳定性(化合物 20f,T1/2 = 249.83 分钟;化合物 20g,T1/2 = 282.60 分钟),在大鼠体内的 PK 曲线也很理想(化合物 20f,T1/2 = 5.16 小时,F = 50.5%;化合物 20g,T1/2 = 7.05 小时,F = 27.3%)。更重要的是,这两种化合物都不会延长小鼠切尾模型(10 毫克/千克,口服)的出血时间。这些结果表明,这些化合物在抗血小板疗法中具有很大的应用潜力。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

CiteScore
11.70
自引率
9.00%
发文量
863
审稿时长
29 days
期刊介绍:
The European Journal of Medicinal Chemistry is a global journal that publishes studies on all aspects of medicinal chemistry. It provides a medium for publication of original papers and also welcomes critical review papers.
A typical paper would report on the organic synthesis, characterization and pharmacological evaluation of compounds. Other topics of interest are drug design, QSAR, molecular modeling, drug-receptor interactions, molecular aspects of drug metabolism, prodrug synthesis and drug targeting. The journal expects manuscripts to present the rational for a study, provide insight into the design of compounds or understanding of mechanism, or clarify the targets.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


