Ionic conduction and cathodic properties of CaMO3 (M=Fe and Mn) electrode materials via molecular dynamics and first-principles simulations
IF 4.3
3区 材料科学
Q2 CHEMISTRY, MULTIDISCIPLINARY
引用次数: 0
Abstract
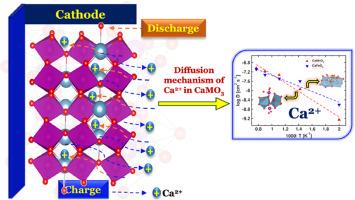
Calcium-ion(Ca-ion) batteries are gaining ever-increasing attention for next-generation energy storage systems due to affordability, highly abundant, high energy density, high theoretical capacity, and low redox potential close to Li-ion. In this work, we deployed the first-principles and classical molecular dynamics simulations to investigate the electronic and diffusive properties of isostructural ternary perovskite CaMO3 (M = Fe and Mn). The transport properties at various temperatures from ion dynamics and electronic properties of CaMO3 perovskites are examined using classical molecular dynamics and quantum mechanical simulations, respectively. We present the microscopic origin of the diffusion of multivalent ions like Ca2+ within the crystal structure of perovskite material and the effects of two transition metals, manganese and iron. Dynamic studies of Ca-ions were performed using molecular dynamic simulation, which depicts the diffusivity and conductivity of Ca-ion in CaMO3 material. We find that the diffusivity in both the crystals increases with temperature; as a result, conductivity increases. Among both the crystals, CaFeO3 requires less activation energy for diffusion and ionic conduction than CaMnO3. Using density functional theory, we calculated specific capacity, electronic density of states, phase stability and equilibrium cell voltage, and charge transfer process during intercalation-deintercalation from first-principles calculations. The electronic behavior of these materials show that CaFeO3 has better electronic and transport properties than CaMnO3.
通过分子动力学和第一原理模拟研究 CaMO3(M=铁和锰)电极材料的离子传导和阴极特性
钙离子(Ca-ion)电池具有价格低廉、资源丰富、能量密度高、理论容量大以及氧化还原电势接近锂离子等优点,因此在下一代储能系统中日益受到关注。在这项工作中,我们利用第一性原理和经典分子动力学模拟研究了等结构三元包晶 CaMO3(M = 铁和锰)的电子和扩散特性。我们利用经典分子动力学和量子力学模拟,分别从离子动力学和 CaMO3 包晶的电子特性出发,研究了其在不同温度下的输运特性。我们介绍了 Ca2+ 等多价离子在包晶材料晶体结构中扩散的微观起源,以及锰和铁这两种过渡金属的影响。利用分子动力学模拟对 Ca 离子进行了动态研究,描绘了 Ca 离子在 CaMO3 材料中的扩散率和导电率。我们发现,两种晶体中的扩散率都会随着温度的升高而增加,因此电导率也会随之增加。在这两种晶体中,CaFeO3 的扩散和离子传导所需的活化能低于 CaMnO3。我们利用密度泛函理论计算了比容量、电子态密度、相稳定性和平衡电池电压,并通过第一原理计算了插层-插层过程中的电荷转移过程。这些材料的电子行为表明,CaFeO3 比 CaMnO3 具有更好的电子和传输特性。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

CiteScore
7.80
自引率
2.50%
发文量
605
审稿时长
40 days
期刊介绍:
The Journal of Physics and Chemistry of Solids is a well-established international medium for publication of archival research in condensed matter and materials sciences. Areas of interest broadly include experimental and theoretical research on electronic, magnetic, spectroscopic and structural properties as well as the statistical mechanics and thermodynamics of materials. The focus is on gaining physical and chemical insight into the properties and potential applications of condensed matter systems.
Within the broad scope of the journal, beyond regular contributions, the editors have identified submissions in the following areas of physics and chemistry of solids to be of special current interest to the journal:
Low-dimensional systems
Exotic states of quantum electron matter including topological phases
Energy conversion and storage
Interfaces, nanoparticles and catalysts.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


