Proteome-scale structural prediction of the giant Marseillevirus reveals conserved folds and putative homologs of the hypothetical proteins
Abstract
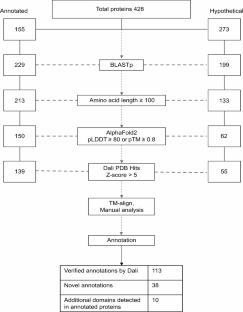
A significant proportion of the highly divergent and novel proteins of giant viruses are termed “hypothetical” due to the absence of detectable homologous sequences in the existing databases. The quality of genome and proteome annotations often relies on the identification of signature sequences and motifs in order to assign putative functions to the gene products. These annotations serve as the first set of information for researchers to develop workable hypotheses for further experimental research. The structure-function relationship of proteins suggests that proteins with similar functions may also exhibit similar folding patterns. Here, we report the first proteome-wide structure prediction of the giant Marseillevirus. We use AlphaFold-predicted structures and their comparative analysis with the experimental structures in the PDB database to preliminarily annotate the viral proteins. Our work highlights the conservation of structural folds in proteins with highly divergent sequences and reveals potentially paralogous relationships among them. We also provide evidence for gene duplication and fusion as contributing factors to giant viral genome expansion and evolution. With the easily accessible AlphaFold and other advanced bioinformatics tools for high-confidence de novo structure prediction, we propose a combined sequence and predicted-structure-based proteome annotation approach for the initial characterization of novel and complex organisms or viruses.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


