CO oxidation on IrO2(110) surfaces
IF 2.1
4区 化学
Q3 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
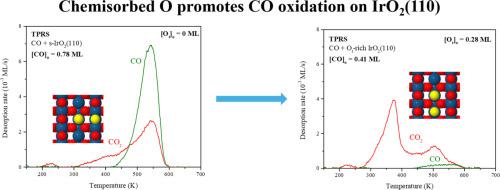
We investigated the oxidation of CO on stoichiometric and O-rich IrO2(110) surfaces using temperature programmed reaction spectroscopy (TPRS), density functional theory (DFT) calculations and microkinetic simulations. Adsorbed CO on the s-IrO2(110) surface generates CO and CO2 peaks near 545 K during TPRS, and only about 38 % of the CO oxidized to CO2 when the initial CO layer was saturated. Pre-adsorbed O-atoms, so-called on-top oxygen (Ot), promote the oxidation of CO adsorbed on IrO2(110). On the Ot-covered surface, CO oxidation by Ot atoms produces a CO2 TPRS peak at ∼370 K, and all of the initially adsorbed CO oxidizes to CO2 when the initial Ot coverage is greater than the CO coverage. In agreement with the TPRS results, DFT calculations predict that the barrier is about 100 kJ/mol lower for CO oxidation by an Ot atom than a lattice O-atom of IrO2(110). A microkinetic model, parameterized with energy barriers computed using DFT, accurately reproduces the CO and CO2 TPRS traces only after CO binding energies are lowered to values determined using a hybrid exchange-correlation functional and the barrier for CO molecules to fill bridging O-vacancies is lowered. The simulations predict that O-vacancies play an important role in mediating the CO oxidation kinetics on s-IrO2(110), and thereby demonstrate the importance of future spectroscopic studies aimed at characterizing the nature of the surface CO and O species involved in reaction. This study provides new insights for understanding CO oxidation on IrO2(110), and provides evidence that several elementary steps can be involved in governing this chemistry.
IrO2(110)表面的 CO 氧化作用
我们利用温度编程反应光谱(TPRS)、密度泛函理论(DFT)计算和微动力学模拟研究了CO在化学计量和富含O的IrO2(110)表面上的氧化过程。在 TPRS 过程中,s-IrO2(110) 表面吸附的 CO 在 545 K 附近产生 CO 和 CO2 峰,当初始 CO 层饱和时,只有约 38% 的 CO 氧化成 CO2。预先吸附的 O 原子,即所谓的顶部氧(Ot),促进了吸附在 IrO2(110)上的 CO 的氧化。在Ot覆盖的表面上,Ot原子对CO的氧化作用会在∼370 K时产生一个CO2 TPRS峰,当初始Ot覆盖率大于CO覆盖率时,所有初始吸附的CO都会氧化成CO2。与 TPRS 结果一致,DFT 计算预测,Ot 原子氧化 CO 的势垒比 IrO2(110) 晶格 O 原子氧化 CO 的势垒低约 100 kJ/mol。只有在一氧化碳结合能降低到使用混合交换相关函数确定的值以及一氧化碳分子填充桥接 O-空位的障碍降低之后,使用 DFT 计算的能垒参数化的微动力学模型才能准确地再现一氧化碳和二氧化碳的 TPRS 轨迹。模拟预测 O-空位在介导 s-IrO2(110)上的 CO 氧化动力学中发挥了重要作用,从而证明了未来旨在确定参与反应的表面 CO 和 O 物种性质的光谱研究的重要性。这项研究为理解二氧化钛(IrO2)(110) 上的一氧化碳氧化作用提供了新的视角,并提供了证据,证明这一化学反应可能涉及几个基本步骤。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Surface Science
化学-物理:凝聚态物理
CiteScore
3.30
自引率
5.30%
发文量
137
审稿时长
25 days
期刊介绍:
Surface Science is devoted to elucidating the fundamental aspects of chemistry and physics occurring at a wide range of surfaces and interfaces and to disseminating this knowledge fast. The journal welcomes a broad spectrum of topics, including but not limited to:
• model systems (e.g. in Ultra High Vacuum) under well-controlled reactive conditions
• nanoscale science and engineering, including manipulation of matter at the atomic/molecular scale and assembly phenomena
• reactivity of surfaces as related to various applied areas including heterogeneous catalysis, chemistry at electrified interfaces, and semiconductors functionalization
• phenomena at interfaces relevant to energy storage and conversion, and fuels production and utilization
• surface reactivity for environmental protection and pollution remediation
• interactions at surfaces of soft matter, including polymers and biomaterials.
Both experimental and theoretical work, including modeling, is within the scope of the journal. Work published in Surface Science reaches a wide readership, from chemistry and physics to biology and materials science and engineering, providing an excellent forum for cross-fertilization of ideas and broad dissemination of scientific discoveries.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


