High-throughput single-cell sequencing of activated sludge microbiome
IF 14.3
1区 环境科学与生态学
Q1 ENVIRONMENTAL SCIENCES
引用次数: 0
Abstract
Wastewater treatment plants (WWTPs) represent one of biotechnology's largest and most critical applications, playing a pivotal role in environmental protection and public health. In WWTPs, activated sludge (AS) plays a major role in removing contaminants and pathogens from wastewater. While metagenomics has advanced our understanding of microbial communities, it still faces challenges in revealing the genomic heterogeneity of cells, uncovering the microbial dark matter, and establishing precise links between genetic elements and their host cells as a bulk method. These issues could be largely resolved by single-cell sequencing, which can offer unprecedented resolution to show the unique genetic information. Here we show the high-throughput single-cell sequencing to the AS microbiome. The single-amplified genomes (SAGs) of 15,110 individual cells were clustered into 2,454 SAG bins. We find that 27.5% of the genomes in the AS microbial community represent potential novel species, highlighting the presence of microbial dark matter. Furthermore, we identified 1,137 antibiotic resistance genes (ARGs), 10,450 plasmid fragments, and 1,343 phage contigs, with shared plasmid and phage groups broadly distributed among hosts, indicating a high frequency of horizontal gene transfer (HGT) within the AS microbiome. Complementary analysis using 1,529 metagenome-assembled genomes from the AS samples allowed for the taxonomic classification of 98 SAG bins, which were previously unclassified. Our study establishes the feasibility of single-cell sequencing in characterizing the AS microbiome, providing novel insights into its ecological dynamics, and deepening our understanding of HGT processes, particularly those involving ARGs. Additionally, this valuable tool could monitor the distribution, spread, and pathogenic hosts of ARGs both within AS environments and between AS and other environments, which will ultimately contribute to developing a health risk evaluation system for diverse environments within a One Health framework.
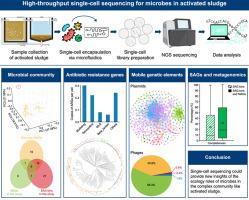
活性污泥微生物组的高通量单细胞测序
污水处理厂(WWTP)是生物技术最大、最关键的应用领域之一,在环境保护和公共卫生方面发挥着举足轻重的作用。在污水处理厂中,活性污泥(AS)在去除废水中的污染物和病原体方面发挥着重要作用。虽然元基因组学促进了我们对微生物群落的了解,但作为一种批量方法,它在揭示细胞基因组异质性、发掘微生物暗物质以及建立遗传元素与其宿主细胞之间的精确联系方面仍面临挑战。单细胞测序可以在很大程度上解决这些问题,因为单细胞测序可以提供前所未有的分辨率来显示独特的遗传信息。在这里,我们展示了对强直性脊柱炎微生物组的高通量单细胞测序。我们将 15110 个单细胞的单扩增基因组(SAG)聚类到 2454 个 SAG bins 中。我们发现,在 AS 微生物群落中,27.5% 的基因组代表了潜在的新物种,凸显了微生物暗物质的存在。此外,我们还发现了1,137个抗生素耐药基因(ARGs)、10,450个质粒片段和1,343个噬菌体等位组,其中共享质粒和噬菌体组广泛分布于宿主之间,这表明强直性脊柱炎微生物群落中水平基因转移(HGT)的频率很高。利用来自强直性脊柱炎样本的1,529个元基因组组装的基因组进行补充分析,可以对98个SAG分区进行分类,而这些分区以前是未分类的。我们的研究证实了单细胞测序在描述强直性脊柱炎微生物组特征方面的可行性,为了解其生态动态提供了新的视角,并加深了我们对HGT过程的理解,特别是那些涉及ARGs的过程。此外,这一宝贵的工具还能监测ARGs在强直性脊柱炎环境中以及强直性脊柱炎与其他环境之间的分布、传播和致病宿主,这最终将有助于在 "一个健康 "框架内为不同环境开发一个健康风险评估系统。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Environmental Science and Ecotechnology
Multiple-
CiteScore
20.40
自引率
6.30%
发文量
11
审稿时长
18 days
期刊介绍:
Environmental Science & Ecotechnology (ESE) is an international, open-access journal publishing original research in environmental science, engineering, ecotechnology, and related fields. Authors publishing in ESE can immediately, permanently, and freely share their work. They have license options and retain copyright. Published by Elsevier, ESE is co-organized by the Chinese Society for Environmental Sciences, Harbin Institute of Technology, and the Chinese Research Academy of Environmental Sciences, under the supervision of the China Association for Science and Technology.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


