{"title":"First-principles calculations to investigate Li segregation to Al2CuMg (001)/Al (021) interface in Al–Cu–Li–Mg alloys","authors":"Jian-Gang Yao, Deng-Feng Yin, Ming-Chun Zhao, Yong Jiang","doi":"10.1140/epjb/s10051-024-00779-0","DOIUrl":null,"url":null,"abstract":"<div><p>In Al–Cu–Li–Mg alloys, Li not only imposes the effects on the precipitation of T1, δ′, θ′, S et al., but also segregates to the S (Al<sub>2</sub>CuMg)/Al interface to stabilize the S precipitates. However, such segregation behavior could not be characterized experimentally at the atomic degree due to the low mass of Li. Also, the fine structure of S/Al interface is not yet clear. This work applied first-principles calculations to study these issues. The numerical values of surface energies for three non-stoichiometric surfaces (Al-1, Al-2, and CuMg terminations) of Al<sub>2</sub>CuMg (001) surface were calculated as ~ 1.3 J/m<sup>2</sup> using a special method. The lowest energy of Al<sub>2</sub>CuMg (001)/Al (021) structure was obtained based on the calculation of energies and interface separated work. The segregation of Li at Al<sub>2</sub>CuMg (001)/Al (021) interface was clarified: (i) Li substituted not only one Al atom, but also all the Al atoms of the ‘3’ layer rather than the ‘1’or ‘2’ layer of the Al matrix side at the interface, (ii) Li that substituted Al atoms of the Al matrix side preferred to play on the role of Mg in Al<sub>2</sub>CuMg, trying to expand Al<sub>2</sub>CuMg bulk to maintain the interface stability, (iii) Li segregation is helpful to improve interface strength. This work provides a considerable insight for Li segregation to Al<sub>2</sub>CuMg (001)/Al (021) interface in Al–Cu–Li–Mg alloys.</p><h3>Graphical Abstract</h3>\n<div><figure><div><div><picture><source><img></source></picture></div></div></figure></div></div>","PeriodicalId":787,"journal":{"name":"The European Physical Journal B","volume":"97 9","pages":""},"PeriodicalIF":1.6000,"publicationDate":"2024-09-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The European Physical Journal B","FirstCategoryId":"4","ListUrlMain":"https://link.springer.com/article/10.1140/epjb/s10051-024-00779-0","RegionNum":4,"RegionCategory":"物理与天体物理","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"PHYSICS, CONDENSED MATTER","Score":null,"Total":0}
引用次数: 0
Abstract
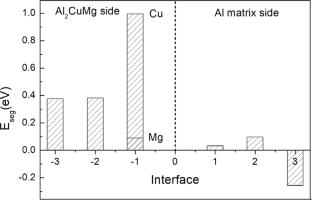
In Al–Cu–Li–Mg alloys, Li not only imposes the effects on the precipitation of T1, δ′, θ′, S et al., but also segregates to the S (Al2CuMg)/Al interface to stabilize the S precipitates. However, such segregation behavior could not be characterized experimentally at the atomic degree due to the low mass of Li. Also, the fine structure of S/Al interface is not yet clear. This work applied first-principles calculations to study these issues. The numerical values of surface energies for three non-stoichiometric surfaces (Al-1, Al-2, and CuMg terminations) of Al2CuMg (001) surface were calculated as ~ 1.3 J/m2 using a special method. The lowest energy of Al2CuMg (001)/Al (021) structure was obtained based on the calculation of energies and interface separated work. The segregation of Li at Al2CuMg (001)/Al (021) interface was clarified: (i) Li substituted not only one Al atom, but also all the Al atoms of the ‘3’ layer rather than the ‘1’or ‘2’ layer of the Al matrix side at the interface, (ii) Li that substituted Al atoms of the Al matrix side preferred to play on the role of Mg in Al2CuMg, trying to expand Al2CuMg bulk to maintain the interface stability, (iii) Li segregation is helpful to improve interface strength. This work provides a considerable insight for Li segregation to Al2CuMg (001)/Al (021) interface in Al–Cu–Li–Mg alloys.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


