{"title":"Quantum Molecular Charge-Transfer Model for Multistep Auger-Meitner Decay Cascade Dynamics.","authors":"Adam E A Fouda, Stephen H Southworth, Phay J Ho","doi":"10.1021/acs.jctc.4c00778","DOIUrl":null,"url":null,"abstract":"<p><p>The fragmentation of molecular cations following inner-shell decay processes in molecules containing heavy elements underpins the X-ray damage effects observed in X-ray scattering measurements of biological and chemical materials, as well as in medical applications involving Auger electron-emitting radionuclides. Traditionally, these processes are modeled using simulations that describe the electronic structure at an atomic level, thereby omitting molecular bonding effects. This work addresses the gap by introducing a novel approach that couples Auger-Meitner decay to nuclear dynamics across multiple decay steps, by developing a decay spawning dynamics algorithm and applying it to potential energy surfaces characterized with <i>ab initio</i> molecular dynamics simulations. We showcase the approach on a model decay cascade following K-shell ionization of IBr and subsequent <i>K</i>β fluorescence decay. We examine two competing channels that undergo two decay steps, resulting in ion pairs with a total 3+ charge state. This approach provides a continuous description of the electron transfer dynamics occurring during the multistep decay cascade and molecular fragmentation, revealing the combined inner-shell decay and charge transfer time scale to be approximately 75 fs. Our computed kinetic energies of ion fragments show good agreement with experimental data.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":" ","pages":"8782-8794"},"PeriodicalIF":5.7000,"publicationDate":"2024-10-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c00778","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/11 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
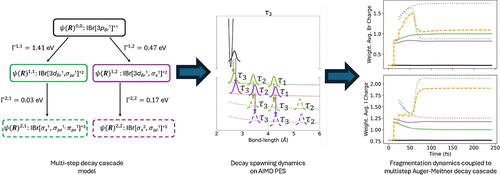
The fragmentation of molecular cations following inner-shell decay processes in molecules containing heavy elements underpins the X-ray damage effects observed in X-ray scattering measurements of biological and chemical materials, as well as in medical applications involving Auger electron-emitting radionuclides. Traditionally, these processes are modeled using simulations that describe the electronic structure at an atomic level, thereby omitting molecular bonding effects. This work addresses the gap by introducing a novel approach that couples Auger-Meitner decay to nuclear dynamics across multiple decay steps, by developing a decay spawning dynamics algorithm and applying it to potential energy surfaces characterized with ab initio molecular dynamics simulations. We showcase the approach on a model decay cascade following K-shell ionization of IBr and subsequent Kβ fluorescence decay. We examine two competing channels that undergo two decay steps, resulting in ion pairs with a total 3+ charge state. This approach provides a continuous description of the electron transfer dynamics occurring during the multistep decay cascade and molecular fragmentation, revealing the combined inner-shell decay and charge transfer time scale to be approximately 75 fs. Our computed kinetic energies of ion fragments show good agreement with experimental data.
在对生物和化学材料进行 X 射线散射测量时,以及在涉及奥杰电子发射放射性核素的医疗应用中,分子阳离子在含有重元素的分子中发生内壳衰变过程后碎裂,这是观察到 X 射线损伤效应的基础。传统上,这些过程都是通过在原子层面描述电子结构的模拟来建模的,因此忽略了分子键效应。这项研究通过开发一种衰变催生动力学算法,并将其应用于以原子分子动力学模拟为特征的势能面,引入了一种将奥杰-迈特纳衰变与多个衰变步骤的核动力学结合起来的新方法,从而弥补了这一空白。我们在 IBr 的 K 壳电离和随后的 Kβ 荧光衰变之后的模型衰变级联上展示了这种方法。我们研究了两个相互竞争的通道,它们经历了两个衰变步骤,产生了总电荷状态为 3+ 的离子对。这种方法连续描述了多步衰变级联和分子破碎过程中发生的电子转移动力学,揭示了内壳衰变和电荷转移的综合时间尺度约为 75 fs。我们计算的离子碎片动能与实验数据显示出良好的一致性。
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


