Human DNA polymerase ε is a source of C>T mutations at CpG dinucleotides
IF 31.7
1区 生物学
Q1 GENETICS & HEREDITY
引用次数: 0
Abstract
C-to-T transitions in CpG dinucleotides are the most prevalent mutations in human cancers and genetic diseases. These mutations have been attributed to deamination of 5-methylcytosine (5mC), an epigenetic modification found on CpGs. We recently linked CpG>TpG mutations to replication and hypothesized that errors introduced by polymerase ε (Pol ε) may represent an alternative source of mutations. Here we present a new method called polymerase error rate sequencing (PER-seq) to measure the error spectrum of DNA polymerases in isolation. We find that the most common human cancer-associated Pol ε mutant (P286R) produces an excess of CpG>TpG errors, phenocopying the mutation spectrum of tumors carrying this mutation and deficiencies in mismatch repair. Notably, we also discover that wild-type Pol ε has a sevenfold higher error rate when replicating 5mCpG compared to C in other contexts. Together, our results from PER-seq and human cancers demonstrate that replication errors are a major contributor to CpG>TpG mutagenesis in replicating cells, fundamentally changing our understanding of this important disease-causing mutational mechanism. A new method called polymerase error rate sequencing (PER-seq) can measure the nucleotide misincorporation rate of DNA polymerases. DNA polymerase ε mutants produce an excess of CpG
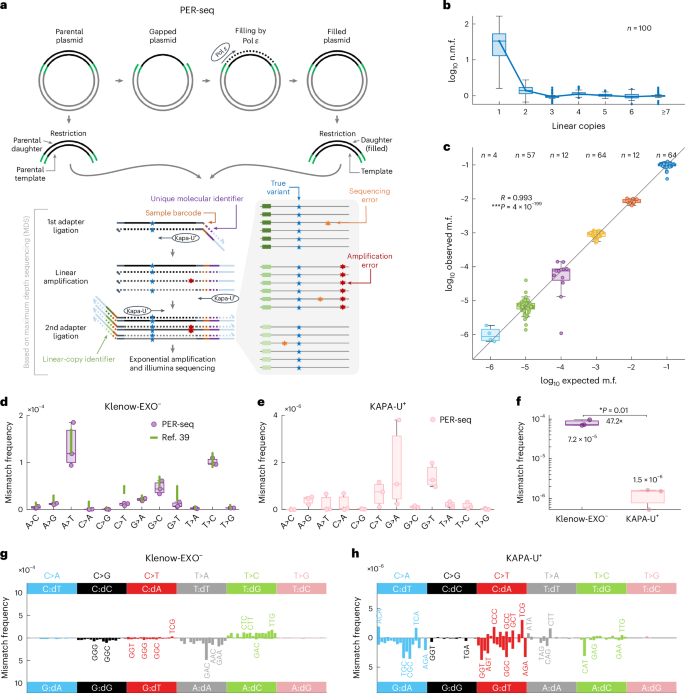
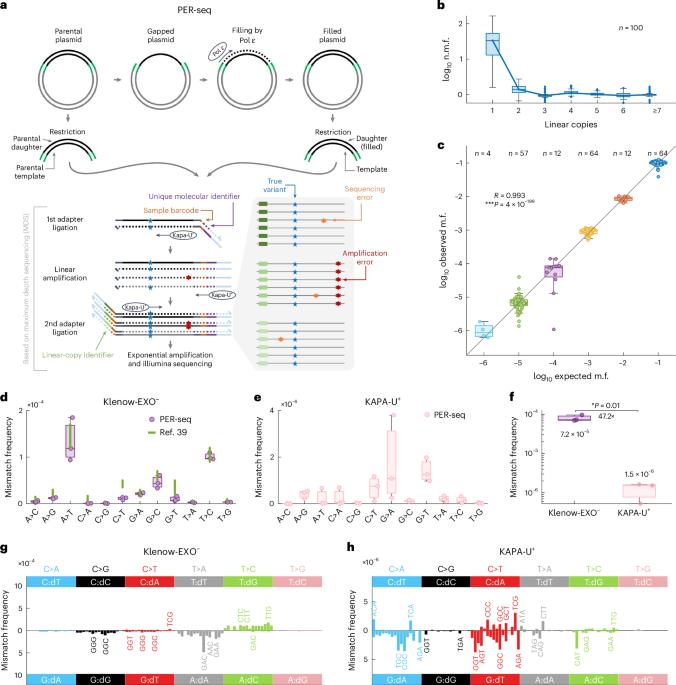
人类 DNA 聚合酶 ε 是 CpG 二核苷酸上 C>T 突变的来源之一
CpG 二核苷酸的 C-T 转换是人类癌症和遗传疾病中最常见的突变。这些突变归因于 5-甲基胞嘧啶(5mC)的脱氨基作用,5-甲基胞嘧啶是 CpGs 上的一种表观遗传修饰。我们最近将 CpG>TpG 突变与复制联系起来,并假设聚合酶 ε(Pol ε)引入的错误可能是突变的另一个来源。在这里,我们提出了一种称为聚合酶错误率测序(PER-seq)的新方法,用于单独测量 DNA 聚合酶的错误谱。我们发现,最常见的人类癌症相关 Pol ε 突变体(P286R)产生了过多的 CpG>TpG 错误,表征了携带这种突变和错配修复缺陷的肿瘤的突变谱。值得注意的是,我们还发现野生型 Pol ε 复制 5mCpG 时的错误率比其他情况下的 C 高七倍。我们从 PER-seq 和人类癌症中获得的结果共同证明,复制错误是复制细胞中 CpG>TpG 诱变的主要因素,从根本上改变了我们对这一重要致病突变机制的认识。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Nature genetics
生物-遗传学
CiteScore
43.00
自引率
2.60%
发文量
241
审稿时长
3 months
期刊介绍:
Nature Genetics publishes the very highest quality research in genetics. It encompasses genetic and functional genomic studies on human and plant traits and on other model organisms. Current emphasis is on the genetic basis for common and complex diseases and on the functional mechanism, architecture and evolution of gene networks, studied by experimental perturbation.
Integrative genetic topics comprise, but are not limited to:
-Genes in the pathology of human disease
-Molecular analysis of simple and complex genetic traits
-Cancer genetics
-Agricultural genomics
-Developmental genetics
-Regulatory variation in gene expression
-Strategies and technologies for extracting function from genomic data
-Pharmacological genomics
-Genome evolution
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


