{"title":"Haploidy-linked cell proliferation defects limit larval growth in zebrafish.","authors":"Kan Yaguchi, Daiki Saito, Triveni Menon, Akira Matsura, Miyu Hosono, Takeomi Mizutani, Tomoya Kotani, Sreelaja Nair, Ryota Uehara","doi":"10.1098/rsob.240126","DOIUrl":null,"url":null,"abstract":"<p><p>Haploid larvae in non-mammalian vertebrates are lethal, with characteristic organ growth retardation collectively called 'haploid syndrome'. In contrast to mammals, whose haploid intolerance is attributed to imprinting misregulation, the cellular principle of haploidy-linked defects in non-mammalian vertebrates remains unknown. Here, we investigated cellular defects that disrupt the ontogeny of gynogenetic haploid zebrafish larvae. Unlike diploid control larvae, haploid larvae manifested unscheduled cell death at the organogenesis stage, attributed to haploidy-linked p53 upregulation. Moreover, we found that haploid larvae specifically suffered the gradual aggravation of mitotic spindle monopolarization during 1-3 days post-fertilization, causing spindle assembly checkpoint-mediated mitotic arrest throughout the entire body. High-resolution imaging revealed that this mitotic defect accompanied the haploidy-linked centrosome loss occurring concomitantly with the gradual decrease in larval cell size. Either resolution of mitotic arrest or depletion of p53 partially improved organ growth in haploid larvae. Based on these results, we propose that haploidy-linked mitotic defects and cell death are parts of critical cellular causes shared among vertebrates that limit the larval growth in the haploid state, contributing to an evolutionary constraint on allowable ploidy status in the vertebrate life cycle.</p>","PeriodicalId":19629,"journal":{"name":"Open Biology","volume":"14 10","pages":"240126"},"PeriodicalIF":3.6000,"publicationDate":"2024-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11461072/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Open Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1098/rsob.240126","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/9 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
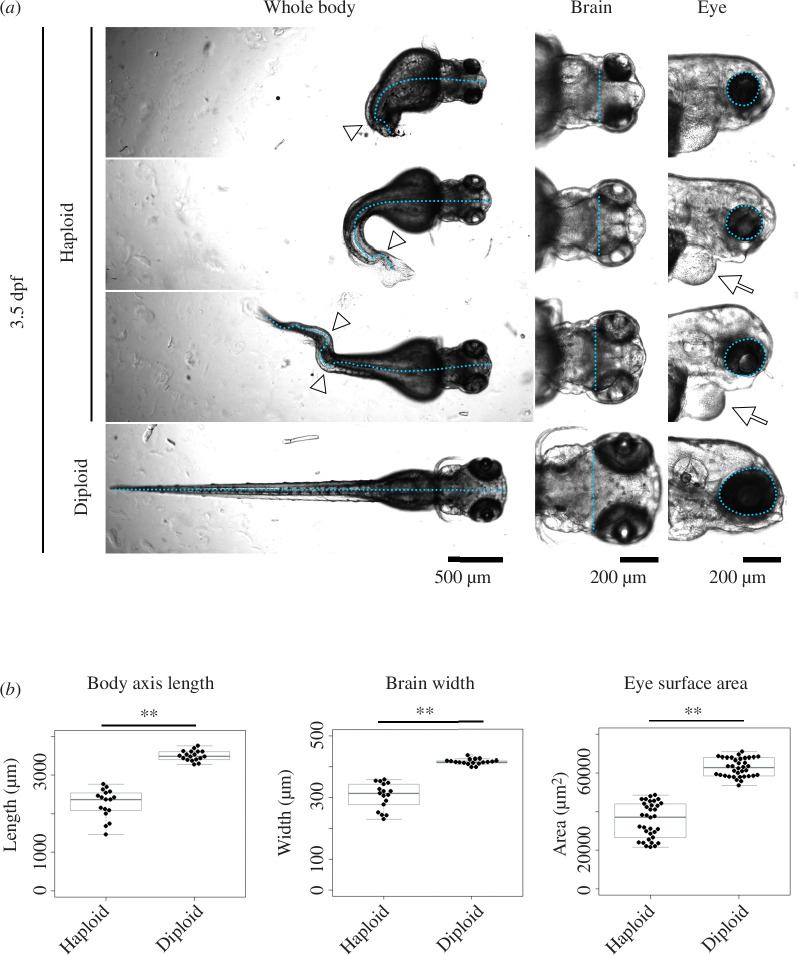
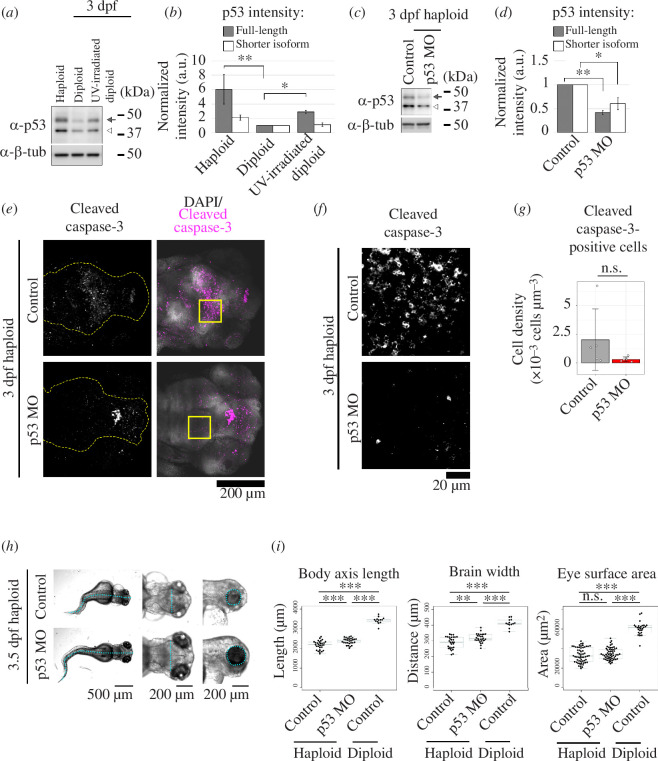
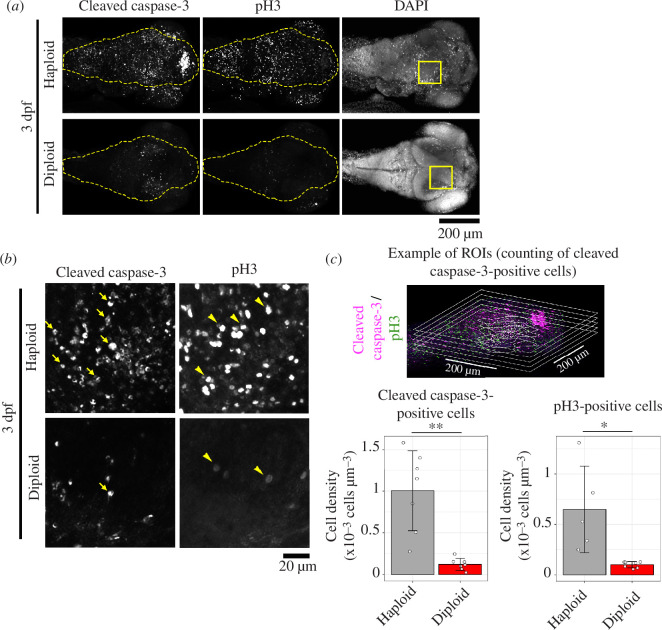
Haploid larvae in non-mammalian vertebrates are lethal, with characteristic organ growth retardation collectively called 'haploid syndrome'. In contrast to mammals, whose haploid intolerance is attributed to imprinting misregulation, the cellular principle of haploidy-linked defects in non-mammalian vertebrates remains unknown. Here, we investigated cellular defects that disrupt the ontogeny of gynogenetic haploid zebrafish larvae. Unlike diploid control larvae, haploid larvae manifested unscheduled cell death at the organogenesis stage, attributed to haploidy-linked p53 upregulation. Moreover, we found that haploid larvae specifically suffered the gradual aggravation of mitotic spindle monopolarization during 1-3 days post-fertilization, causing spindle assembly checkpoint-mediated mitotic arrest throughout the entire body. High-resolution imaging revealed that this mitotic defect accompanied the haploidy-linked centrosome loss occurring concomitantly with the gradual decrease in larval cell size. Either resolution of mitotic arrest or depletion of p53 partially improved organ growth in haploid larvae. Based on these results, we propose that haploidy-linked mitotic defects and cell death are parts of critical cellular causes shared among vertebrates that limit the larval growth in the haploid state, contributing to an evolutionary constraint on allowable ploidy status in the vertebrate life cycle.
期刊介绍:
Open Biology is an online journal that welcomes original, high impact research in cell and developmental biology, molecular and structural biology, biochemistry, neuroscience, immunology, microbiology and genetics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


