Second-Order Mass-Weighting Scheme for Atom-Centered Density Matrix Propagation Molecular Dynamics
IF 5.7
1区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
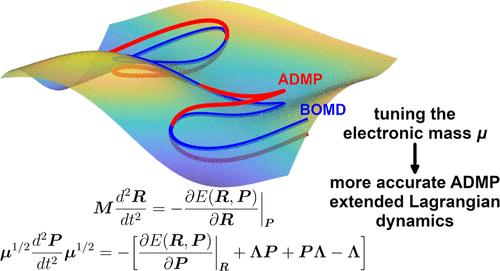
The atom-centered density matrix propagation (ADMP) method is an extended Lagrangian approach to ab initio molecular dynamics, which includes the density matrix in an orthonormalized atom-centered Gaussian basis as additional, fictitious, electronic degrees of freedom, classically propagated along with the nuclear ones. A high adiabaticity between the nuclear and electronic subsystems is mandatory in order to keep the trajectory close to the Born–Oppenheimer (BO) surface. In this regard, the fictitious electronic mass μ, being a symmetric, nondiagonal matrix in its most general form, represents a free parameter, exploitable to optimize the propagation of the electronic density. Although mass-weighting schemes in ADMP exist, a systematic procedure to define an optimal value of the fictitious masses is not available yet. In this work, in order to rationally evaluate the electronic mass, fictitious electronic normal modes are defined through the diagonalization of the Hessian of the electronic density matrix. If the same frequency is imposed on all such modes (compatible with the chosen integration time step), then the corresponding μ matrix can be calculated and then employed for the following propagation. Analysis of several ADMP test simulations reveals that such Hessian-based mass-weighting approach is able to ensure, together with a 0.1/0.2 fs time steps, a high separation between the (real) nuclear and the (fictitious) electronic frequencies, which determines a high adiabaticity. This high, unprecedented, accuracy in the propagation leads, in turn, to low errors in the estimated nuclear vibrational frequencies, making the ADMP method totally comparable to a fully converged BO molecular dynamics simulation but more computationally efficient. This work, therefore, contributes to a further development of the ADMP ab initio molecular dynamics method, aimed at improving its accuracy through a more rational evaluation of the fictitious electronic mass parameter.
原子中心密度矩阵传播分子动力学的二阶质量权重方案
以原子为中心的密度矩阵传播(ADMP)方法是一种扩展的拉格朗日方法,用于原子分子动力学(ab initio molecular dynamics),它将正交化以原子为中心的高斯基中的密度矩阵作为额外的、虚构的电子自由度,与核自由度一起进行经典传播。核子系统和电子子系统之间的高度绝热性是保持轨迹接近玻恩-奥本海默(BO)表面的必要条件。在这方面,虚构电子质量 μ 是一个对称的非对角矩阵,其最一般的形式代表了一个自由参数,可用于优化电子密度的传播。虽然 ADMP 中存在质量加权方案,但目前还没有一个系统的程序来定义虚构质量的最优值。在这项工作中,为了合理地评估电子质量,通过对电子密度矩阵的 Hessian 进行对角化来定义虚构电子法向模态。如果对所有这些模式施加相同的频率(与所选的积分时间步长相匹配),那么就可以计算出相应的 μ 矩阵,然后用于接下来的传播。对多个 ADMP 试验模拟的分析表明,这种基于赫塞斯的质量加权方法能够确保(真实的)核频率和(虚构的)电子频率之间的高分离度,同时还能确保 0.1/0.2 fs 的时间步长,这就决定了高绝热性。这种前所未有的高传播精度反过来又导致了核振动频率估算的低误差,使得 ADMP 方法完全可以与完全收敛的 BO 分子动力学模拟相媲美,但计算效率更高。因此,这项工作有助于进一步发展 ADMP 原子核分子动力学方法,旨在通过更合理地评估虚构电子质量参数来提高其准确性。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of Chemical Theory and Computation
化学-物理:原子、分子和化学物理
CiteScore
9.90
自引率
16.40%
发文量
568
审稿时长
1 months
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


