AAV gene therapy for Duchenne muscular dystrophy: the EMBARK phase 3 randomized trial
IF 58.7
1区 医学
Q1 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 0
Abstract
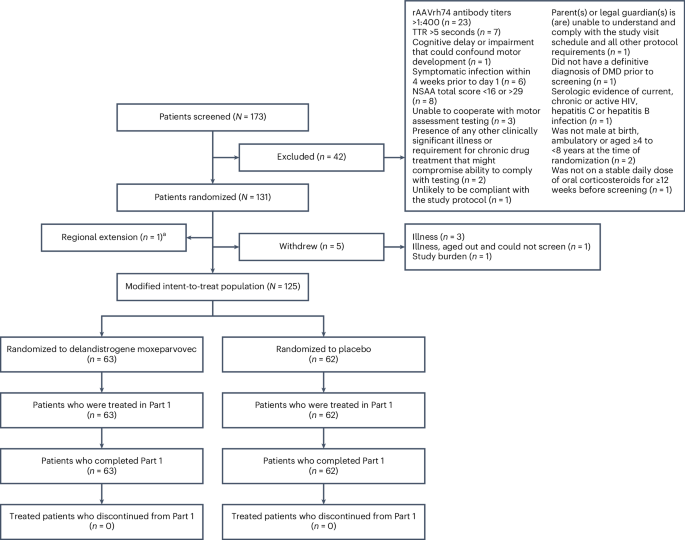
Duchenne muscular dystrophy (DMD) is a rare, X-linked neuromuscular disease caused by pathogenic variants in the DMD gene that result in the absence of functional dystrophin, beginning at birth and leading to progressive impaired motor function, loss of ambulation and life-threatening cardiorespiratory complications. Delandistrogene moxeparvovec, an adeno-associated rh74-viral vector-based gene therapy, addresses absent functional dystrophin in DMD. Here the phase 3 EMBARK study aimed to assess the efficacy and safety of delandistrogene moxeparvovec in patients with DMD. Ambulatory males with DMD, ≥4 years to <8 years of age, were randomized and stratified by age group and North Star Ambulatory Assessment (NSAA) score to single-administration intravenous delandistrogene moxeparvovec (1.33 × 1014 vector genomes per kilogram; n = 63) or placebo (n = 62). At week 52, the primary endpoint, change from baseline in NSAA score, was not met (least squares mean 2.57 (delandistrogene moxeparvovec) versus 1.92 (placebo) points; between-group difference, 0.65; 95% confidence interval (CI), −0.45, 1.74; P = 0.2441). Secondary efficacy endpoints included mean micro-dystrophin expression at week 12: 34.29% (treated) versus 0.00% (placebo). Other secondary efficacy endpoints at week 52 (between-group differences (95% CI)) included: Time to Rise (−0.64 (−1.06, −0.23)), 10-meter Walk/Run (−0.42 (−0.71, −0.13)), stride velocity 95th centile (0.10 (0.00, 0.19)), 100-meter Walk/Run (−3.29 (−8.28, 1.70)), time to ascend 4 steps (–0.36 (−0.71, −0.01)), PROMIS Mobility and Upper Extremity (0.05 (−0.08, 0.19); −0.04 (−0.24, 0.17)) and number of NSAA skills gained/improved (0.19 (−0.67, 1.06)). In total, 674 adverse events were recorded with delandistrogene moxeparvovec and 514 with placebo. There were no deaths, discontinuations or clinically significant complement-mediated adverse events; 7 patients (11.1%) experienced 10 treatment-related serious adverse events. Delandistrogene moxeparvovec did not lead to a significant improvement in NSAA score at week 52. Some of the secondary endpoints numerically favored treatment, although no statistical significance can be claimed. Safety was manageable and consistent with previous delandistrogene moxeparvovec trials. ClinicalTrials.gov: NCT05096221 The primary report of the EMBARK phase 3 trial, testing the AAV-based gene therapy delandistrogene moxeparvovec in Duchenne muscular dystrophy, did not meet its primary endpoint of improvement in NSAA mobility scores compared to placebo. Secondary endpoints show that the therapy was safe and associated with improvements in micro-dystrophin expression and in individual mobility scores.

杜氏肌营养不良症的 AAV 基因疗法:EMBARK 3 期随机试验
杜兴氏肌营养不良症(DMD)是一种罕见的 X 连锁神经肌肉疾病,由 DMD 基因中的致病变体引起,导致功能性肌营养不良蛋白缺失,从出生开始就会导致进行性运动功能受损、丧失行动能力和危及生命的心肺并发症。Delandistrogene moxeparvovec是一种基于腺相关rh74病毒载体的基因疗法,可治疗DMD中缺乏功能性肌营养不良症。EMBARK 3 期研究旨在评估 delandistrogene moxeparvovec 对 DMD 患者的疗效和安全性。对年龄≥4岁至8岁的DMD男性患者进行了随机分组,并按年龄组和North Star Ambulatory Assessment (NSAA)评分进行了分层,分别给予单剂量静脉注射delandistrogene moxeparvovec(每公斤1.33×1014个载体基因组;n = 63)或安慰剂(n = 62)。第52周时,主要终点(NSAA评分与基线相比的变化)未达标(最小二乘法平均值为2.57分(delandistrogene moxeparvovec)对1.92分(安慰剂);组间差异为0.65;95%置信区间(CI)为-0.45,1.74;P=0.2441)。次要疗效终点包括第12周时微量肌营养不良蛋白的平均表达量:34.29%(治疗)对0.00%(安慰剂)。第52周的其他次要疗效终点(组间差异(95% CI))包括起立时间 (-0.64 (-1.06, -0.23))、10 米步行/跑步 (-0.42 (-0.71, -0.13))、步速第 95 百分位数 (0.10 (0.00, 0.19))、100 米步行/跑步 (-3.29 (-8.28, 1. 70))、上升至第 4 百分位数的时间 (0.10 (0.00, 0.19))。70)、上 4 级台阶的时间 (-0.36 (-0.71, -0.01))、PROMIS 移动能力和上肢 (0.05 (-0.08, 0.19); -0.04 (-0.24, 0.17)) 和获得/提高的 NSAA 技能数量 (0.19 (-0.67, 1.06))。使用delandistrogene moxeparvovec和安慰剂分别共记录到674例和514例不良事件。没有出现死亡、停药或有临床意义的补体介导的不良事件;7 名患者(11.1%)出现了 10 次与治疗相关的严重不良事件。第52周时,Delandistrogene moxeparvovec并未显著改善NSAA评分。一些次要终点在数字上有利于治疗,但并不具有统计学意义。安全性尚可,与之前的delandistrogene moxeparvovec试验结果一致。ClinicalTrials.gov:NCT05096221
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Nature Medicine
医学-生化与分子生物学
CiteScore
100.90
自引率
0.70%
发文量
525
审稿时长
1 months
期刊介绍:
Nature Medicine is a monthly journal publishing original peer-reviewed research in all areas of medicine. The publication focuses on originality, timeliness, interdisciplinary interest, and the impact on improving human health. In addition to research articles, Nature Medicine also publishes commissioned content such as News, Reviews, and Perspectives. This content aims to provide context for the latest advances in translational and clinical research, reaching a wide audience of M.D. and Ph.D. readers. All editorial decisions for the journal are made by a team of full-time professional editors.
Nature Medicine consider all types of clinical research, including:
-Case-reports and small case series
-Clinical trials, whether phase 1, 2, 3 or 4
-Observational studies
-Meta-analyses
-Biomarker studies
-Public and global health studies
Nature Medicine is also committed to facilitating communication between translational and clinical researchers. As such, we consider “hybrid” studies with preclinical and translational findings reported alongside data from clinical studies.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


