A Deep Learning-Driven Sampling Technique to Explore the Phase Space of an RNA Stem-Loop
IF 5.7
1区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
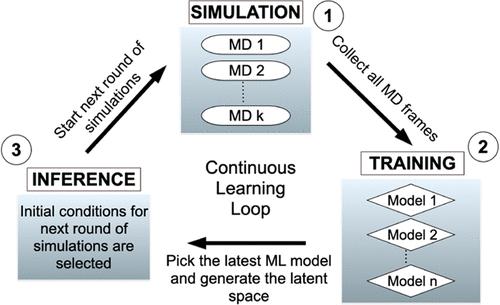
The folding and unfolding of RNA stem-loops are critical biological processes; however, their computational studies are often hampered by the ruggedness of their folding landscape, necessitating long simulation times at the atomistic scale. Here, we adapted DeepDriveMD (DDMD), an advanced deep learning-driven sampling technique originally developed for protein folding, to address the challenges of RNA stem-loop folding. Although tempering- and order parameter-based techniques are commonly used for similar rare-event problems, the computational costs or the need for a priori knowledge about the system often present a challenge in their effective use. DDMD overcomes these challenges by adaptively learning from an ensemble of running MD simulations using generic contact maps as the raw input. DeepDriveMD enables on-the-fly learning of a low-dimensional latent representation and guides the simulation toward the undersampled regions while optimizing the resources to explore the relevant parts of the phase space. We showed that DDMD estimates the free energy landscape of the RNA stem-loop reasonably well at room temperature. Our simulation framework runs at a constant temperature without external biasing potential, hence preserving the information on transition rates, with a computational cost much lower than that of the simulations performed with external biasing potentials. We also introduced a reweighting strategy for obtaining unbiased free energy surfaces and presented a qualitative analysis of the latent space. This analysis showed that the latent space captures the relevant slow degrees of freedom for the RNA folding problem of interest. Finally, throughout the manuscript, we outlined how different parameters are selected and optimized to adapt DDMD for this system. We believe this compendium of decision-making processes will help new users adapt this technique for the rare-event sampling problems of their interest.
探索 RNA 干环相位空间的深度学习驱动采样技术
RNA 干环的折叠和展开是关键的生物过程;然而,由于其折叠景观崎岖不平,需要在原子尺度上进行长时间的模拟,其计算研究往往受到阻碍。在这里,我们将最初为蛋白质折叠开发的先进深度学习驱动采样技术 DeepDriveMD (DDMD) 用于应对 RNA 干环折叠的挑战。虽然基于节制和阶次参数的技术常用于类似的罕见事件问题,但计算成本或对系统先验知识的需求往往对它们的有效使用构成挑战。DDMD 克服了这些挑战,它使用通用接触图作为原始输入,从运行的 MD 仿真集合中进行自适应学习。DeepDriveMD 实现了对低维潜在表示的即时学习,并在优化资源以探索相空间相关部分的同时,引导模拟向未充分采样的区域发展。我们的研究表明,DDMD 能在室温下合理地估算出 RNA 干环的自由能谱。我们的模拟框架在恒定温度下运行,不需要外部偏置电势,因此保留了过渡率的信息,计算成本远远低于使用外部偏置电势进行的模拟。我们还引入了一种用于获得无偏自由能表面的重新加权策略,并对潜能空间进行了定性分析。分析表明,潜空间捕捉到了 RNA 折叠问题的相关慢自由度。最后,在整个手稿中,我们概述了如何选择和优化不同参数,使 DDMD 适用于该系统。我们相信,这份决策过程汇编将帮助新用户调整这项技术,以解决他们感兴趣的罕见事件采样问题。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of Chemical Theory and Computation
化学-物理:原子、分子和化学物理
CiteScore
9.90
自引率
16.40%
发文量
568
审稿时长
1 months
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


