Periodic Local Coupled-Cluster Theory for Insulators and Metals
IF 5.7
1区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
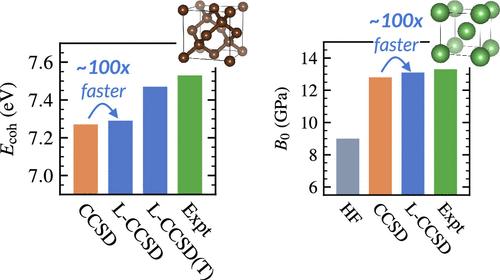
We describe the implementation details of periodic local coupled-cluster theory with single and double excitations (CCSD) and perturbative triple excitations [CCSD(T)] using local natural orbitals (LNOs) and k-point symmetry. We discuss and compare several choices for orbital localization, fragmentation, and LNO construction. By studying diamond and lithium, we demonstrate that periodic LNO-CC theory can be applied with equal success to both insulators and metals, achieving speedups of 2 to 3 orders of magnitude even for moderately sized k-point meshes. Our final predictions of the equilibrium cohesive energy, lattice constant, and bulk modulus for diamond and lithium are in good agreement with previous theoretical predictions and experimental results.
绝缘体和金属的周期局部耦合簇理论
我们描述了使用局部自然轨道(LNO)和 k 点对称的周期性局部耦合簇理论的实施细节,包括单激和双激(CCSD)以及微扰三激(CCSD(T))。我们讨论并比较了轨道定位、碎裂和 LNO 构建的几种选择。通过对金刚石和锂的研究,我们证明了周期性 LNO-CC 理论可以同样成功地应用于绝缘体和金属,即使对于中等大小的 k 点网格,也能实现 2 到 3 个数量级的提速。我们对金刚石和锂的平衡内聚能、晶格常数和体积模量的最终预测与之前的理论预测和实验结果非常吻合。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of Chemical Theory and Computation
化学-物理:原子、分子和化学物理
CiteScore
9.90
自引率
16.40%
发文量
568
审稿时长
1 months
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


