Very-Large-Scale GPU-Accelerated Nuclear Gradient of Time-Dependent Density Functional Theory with Tamm-Dancoff Approximation and Range-Separated Hybrid Functionals.
Inkoo Kim, Daun Jeong, Leah P Weisburn, Alexandra Alexiu, Troy Van Voorhis, Young Min Rhee, Won-Joon Son, Hyung-Jin Kim, Jinkyu Yim, Sungmin Kim, Yeonchoo Cho, Inkook Jang, Seungmin Lee, Dae Sin Kim
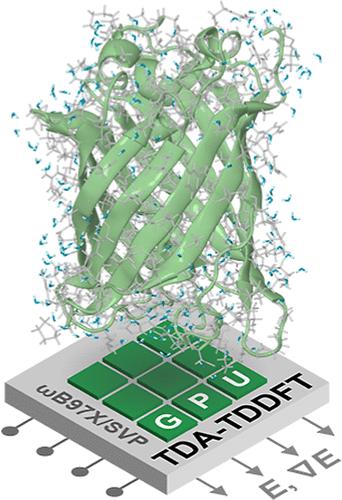
{"title":"Very-Large-Scale GPU-Accelerated Nuclear Gradient of Time-Dependent Density Functional Theory with Tamm-Dancoff Approximation and Range-Separated Hybrid Functionals.","authors":"Inkoo Kim, Daun Jeong, Leah P Weisburn, Alexandra Alexiu, Troy Van Voorhis, Young Min Rhee, Won-Joon Son, Hyung-Jin Kim, Jinkyu Yim, Sungmin Kim, Yeonchoo Cho, Inkook Jang, Seungmin Lee, Dae Sin Kim","doi":"10.1021/acs.jctc.4c01003","DOIUrl":null,"url":null,"abstract":"<p><p>Modern graphics processing units (GPUs) provide an unprecedented level of computing power. In this study, we present a high-performance, multi-GPU implementation of the analytical nuclear gradient for Kohn-Sham time-dependent density functional theory (TDDFT), employing the Tamm-Dancoff approximation (TDA) and Gaussian-type atomic orbitals as basis functions. We discuss GPU-efficient algorithms for the derivatives of electron repulsion integrals and exchange-correlation functionals within the range-separated scheme. As an illustrative example, we calculate the TDA-TDDFT gradient of the S<sub>1</sub> state of a full-scale green fluorescent protein with explicit water solvent molecules, totaling 4353 atoms, at the ωB97X/def2-SVP level of theory. Our algorithm demonstrates favorable parallel efficiencies on a high-speed distributed system equipped with 256 Nvidia A100 GPUs, achieving >70% with up to 64 GPUs and 31% with 256 GPUs, effectively leveraging the capabilities of modern high-performance computing systems.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":null,"pages":null},"PeriodicalIF":5.7000,"publicationDate":"2024-10-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c01003","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/7 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
Modern graphics processing units (GPUs) provide an unprecedented level of computing power. In this study, we present a high-performance, multi-GPU implementation of the analytical nuclear gradient for Kohn-Sham time-dependent density functional theory (TDDFT), employing the Tamm-Dancoff approximation (TDA) and Gaussian-type atomic orbitals as basis functions. We discuss GPU-efficient algorithms for the derivatives of electron repulsion integrals and exchange-correlation functionals within the range-separated scheme. As an illustrative example, we calculate the TDA-TDDFT gradient of the S1 state of a full-scale green fluorescent protein with explicit water solvent molecules, totaling 4353 atoms, at the ωB97X/def2-SVP level of theory. Our algorithm demonstrates favorable parallel efficiencies on a high-speed distributed system equipped with 256 Nvidia A100 GPUs, achieving >70% with up to 64 GPUs and 31% with 256 GPUs, effectively leveraging the capabilities of modern high-performance computing systems.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


