Yuhan Zheng, Zhijie Sun, Jie Liu, Yi Fan, Zhenyu Li, Jinlong Yang
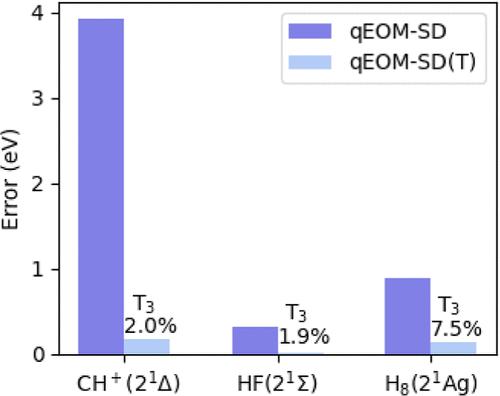
{"title":"Quantum Equation-of-Motion Method with Single, Double, and Triple Excitations.","authors":"Yuhan Zheng, Zhijie Sun, Jie Liu, Yi Fan, Zhenyu Li, Jinlong Yang","doi":"10.1021/acs.jctc.4c01071","DOIUrl":null,"url":null,"abstract":"<p><p>The quantum equation-of-motion (qEOM) method with single and double excitations (qEOM-SD) has been proposed to study electronically excited states, but it fails to handle states dominated by double excitations. In this work, we reformulate the qEOM method within the effective Hamiltonian framework that satisfies the killer condition, and then present an efficient implementation incorporating single, double, and triple excitations. To reduce computational complexity, we employ point-group symmetry and perturbation theory to screen triple excitations, effectively reducing the scaling from <i>N</i><sub>o</sub><sup>6</sup><i>N</i><sub>v</sub><sup>6</sup> to <i>N</i><sub>o</sub><sup>5</sup><i>N</i><sub>v</sub><sup>5</sup>, where <i>N</i><sub>o</sub> and <i>N</i><sub>v</sub> are the numbers of occupied and virtual spin orbitals, respectively. Furthermore, we account for the effect of neglected triple excitations by introducing a perturbative correction to the excitation energy. We apply this method to challenging cases where the qEOM-SD method exhibits significant errors, such as the 2 <sup>1</sup>Δ state of CH<sup>+</sup> and the 2 <sup>1</sup>Σ state of HF. Our new method achieves energy errors below 0.18 eV while incorporating less than 8.2% of triple excitations. Additionally, we extend the operator screening technique to the quantum subspace expansion method for the efficient inclusion of selected triple excitations.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":" ","pages":"9032-9040"},"PeriodicalIF":5.7000,"publicationDate":"2024-10-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c01071","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/7 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
The quantum equation-of-motion (qEOM) method with single and double excitations (qEOM-SD) has been proposed to study electronically excited states, but it fails to handle states dominated by double excitations. In this work, we reformulate the qEOM method within the effective Hamiltonian framework that satisfies the killer condition, and then present an efficient implementation incorporating single, double, and triple excitations. To reduce computational complexity, we employ point-group symmetry and perturbation theory to screen triple excitations, effectively reducing the scaling from No6Nv6 to No5Nv5, where No and Nv are the numbers of occupied and virtual spin orbitals, respectively. Furthermore, we account for the effect of neglected triple excitations by introducing a perturbative correction to the excitation energy. We apply this method to challenging cases where the qEOM-SD method exhibits significant errors, such as the 2 1Δ state of CH+ and the 2 1Σ state of HF. Our new method achieves energy errors below 0.18 eV while incorporating less than 8.2% of triple excitations. Additionally, we extend the operator screening technique to the quantum subspace expansion method for the efficient inclusion of selected triple excitations.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


