Tomasz K Piskorz, Bernadette Lee, Shaoqi Zhan, Fernanda Duarte
{"title":"<i>Metallicious</i>: Automated Force-Field Parameterization of Covalently Bound Metals for Supramolecular Structures.","authors":"Tomasz K Piskorz, Bernadette Lee, Shaoqi Zhan, Fernanda Duarte","doi":"10.1021/acs.jctc.4c00850","DOIUrl":null,"url":null,"abstract":"<p><p>Metal ions play a central, functional, and structural role in many molecular structures, from small catalysts to metal-organic frameworks (MOFs) and proteins. Computational studies of these systems typically employ classical or quantum mechanical approaches or a combination of both. Among classical models, only the covalent metal model reproduces both geometries and charge transfer effects but requires time-consuming parameterization, especially for supramolecular systems containing repetitive units. To streamline this process, we introduce <i>metallicious</i>, a Python tool designed for efficient force-field parameterization of supramolecular structures. <i>Metallicious</i> has been tested on diverse systems including supramolecular cages, knots, and MOFs. Our benchmarks demonstrate that parameters accurately reproduce the reference properties obtained from quantum calculations and crystal structures. Molecular dynamics simulations of the generated structures consistently yield stable simulations in explicit solvent, in contrast to similar simulations performed with nonbonded and cationic dummy models. Overall, <i>metallicious</i> facilitates the atomistic modeling of supramolecular systems, key for understanding their dynamic properties and host-guest interactions. The tool is freely available on GitHub (https://github.com/duartegroup/metallicious).</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":" ","pages":"9060-9071"},"PeriodicalIF":5.7000,"publicationDate":"2024-10-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11500408/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c00850","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/7 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
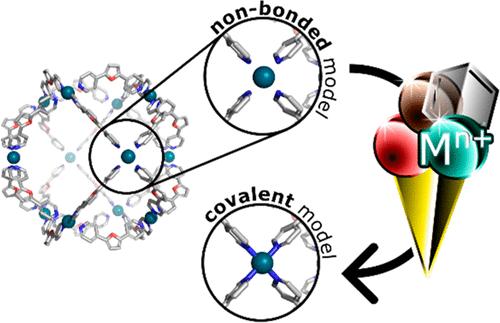
Metal ions play a central, functional, and structural role in many molecular structures, from small catalysts to metal-organic frameworks (MOFs) and proteins. Computational studies of these systems typically employ classical or quantum mechanical approaches or a combination of both. Among classical models, only the covalent metal model reproduces both geometries and charge transfer effects but requires time-consuming parameterization, especially for supramolecular systems containing repetitive units. To streamline this process, we introduce metallicious, a Python tool designed for efficient force-field parameterization of supramolecular structures. Metallicious has been tested on diverse systems including supramolecular cages, knots, and MOFs. Our benchmarks demonstrate that parameters accurately reproduce the reference properties obtained from quantum calculations and crystal structures. Molecular dynamics simulations of the generated structures consistently yield stable simulations in explicit solvent, in contrast to similar simulations performed with nonbonded and cationic dummy models. Overall, metallicious facilitates the atomistic modeling of supramolecular systems, key for understanding their dynamic properties and host-guest interactions. The tool is freely available on GitHub (https://github.com/duartegroup/metallicious).
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


