Timothy D Loose, Patrick G Sahrmann, Thomas S Qu, Gregory A Voth
{"title":"Changing Your Martini Can Still Give You a Hangover.","authors":"Timothy D Loose, Patrick G Sahrmann, Thomas S Qu, Gregory A Voth","doi":"10.1021/acs.jctc.4c00868","DOIUrl":null,"url":null,"abstract":"<p><p>The Martini 3.0 coarse-grained force field, which was parametrized to better capture transferability in top-down coarse-grained models, is analyzed to assess its accuracy in representing thermodynamic and structural properties with respect to the underlying atomistic representation of the system. These results are compared to those obtained following the principles of statistical mechanics that start from the same underlying atomistic system. To this end, the potentials of mean force for lateral association in Martini 3.0 binary lipid bilayers are decomposed into their entropic and enthalpic components and compared to those of corresponding atomistic bilayers that have been projected onto equivalent coarse-grained mappings but evolved under the fully atomistic forces. This is accomplished by applying the reversible work theorem to lateral pair correlation functions between coarse-grained lipid beads taken at a range of different temperatures. The entropy-enthalpy decompositions provide a metric by which the underlying statistical mechanical properties of Martini can be investigated. Overall, Martini 3.0 is found to fail to properly partition entropy and enthalpy for the PMFs compared to the mapped all-atom results, despite changes made to the force field from the Martini 2.0 version. This outcome points to the fact that the development of more accurate top-down coarse-grained models such as Martini will likely necessitate temperature-dependent terms in the corresponding CG force-field; although necessary, this may not be sufficient to improve Martini. In addition to the entropy-enthalpy decompositions, Martini 3.0 produces an incorrect undulation spectrum, in particular at intermediate length scales of biophysical pertinence.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":" ","pages":"9190-9208"},"PeriodicalIF":5.5000,"publicationDate":"2024-10-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11500708/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c00868","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/3 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
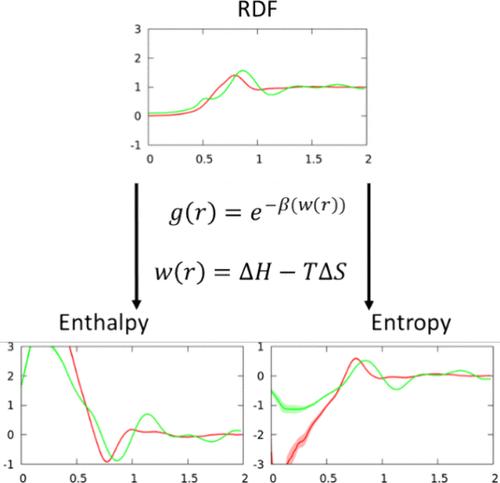
The Martini 3.0 coarse-grained force field, which was parametrized to better capture transferability in top-down coarse-grained models, is analyzed to assess its accuracy in representing thermodynamic and structural properties with respect to the underlying atomistic representation of the system. These results are compared to those obtained following the principles of statistical mechanics that start from the same underlying atomistic system. To this end, the potentials of mean force for lateral association in Martini 3.0 binary lipid bilayers are decomposed into their entropic and enthalpic components and compared to those of corresponding atomistic bilayers that have been projected onto equivalent coarse-grained mappings but evolved under the fully atomistic forces. This is accomplished by applying the reversible work theorem to lateral pair correlation functions between coarse-grained lipid beads taken at a range of different temperatures. The entropy-enthalpy decompositions provide a metric by which the underlying statistical mechanical properties of Martini can be investigated. Overall, Martini 3.0 is found to fail to properly partition entropy and enthalpy for the PMFs compared to the mapped all-atom results, despite changes made to the force field from the Martini 2.0 version. This outcome points to the fact that the development of more accurate top-down coarse-grained models such as Martini will likely necessitate temperature-dependent terms in the corresponding CG force-field; although necessary, this may not be sufficient to improve Martini. In addition to the entropy-enthalpy decompositions, Martini 3.0 produces an incorrect undulation spectrum, in particular at intermediate length scales of biophysical pertinence.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


