Strainy: phasing and assembly of strain haplotypes from long-read metagenome sequencing
IF 32.1
1区 生物学
Q1 BIOCHEMICAL RESEARCH METHODS
引用次数: 0
Abstract
Bacterial species in microbial communities are often represented by mixtures of strains, distinguished by small variations in their genomes. Short-read approaches can be used to detect small-scale variation between strains but fail to phase these variants into contiguous haplotypes. Long-read metagenome assemblers can generate contiguous bacterial chromosomes but often suppress strain-level variation in favor of species-level consensus. Here we present Strainy, an algorithm for strain-level metagenome assembly and phasing from Nanopore and PacBio reads. Strainy takes a de novo metagenomic assembly as input and identifies strain variants, which are then phased and assembled into contiguous haplotypes. Using simulated and mock Nanopore and PacBio metagenome data, we show that Strainy assembles accurate and complete strain haplotypes, outperforming current Nanopore-based methods and comparable with PacBio-based algorithms in completeness and accuracy. We then use Strainy to assemble strain haplotypes of a complex environmental metagenome, revealing distinct strain distribution and mutational patterns in bacterial species. This work presents Strainy, a long-read metagenome assembler that allows the identification of strain distributions and mutational patterns in environmental metagenomes.
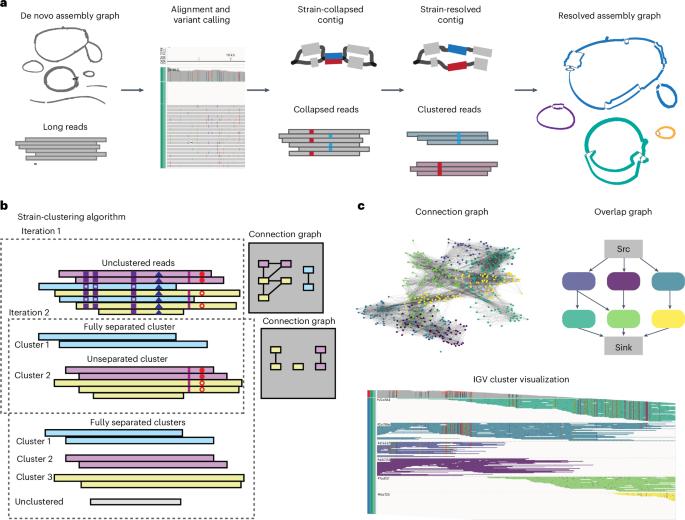
菌株:长线程元基因组测序中菌株单倍型的分期和组装。
微生物群落中的细菌物种通常由菌株混合物代表,以其基因组中的微小变异加以区分。短读数方法可用于检测菌株之间的小规模变异,但无法将这些变异分期为连续的单倍型。长读数元基因组组装器可以生成连续的细菌染色体,但往往会抑制菌株级变异,而倾向于物种级共识。在此,我们介绍 Strainy,这是一种利用 Nanopore 和 PacBio 读数进行菌株级元基因组组装和分期的算法。Strainy 将全新的元基因组组装作为输入,识别菌株变异,然后将其分期并组装成连续的单倍型。我们使用模拟和仿真的 Nanopore 和 PacBio 元基因组数据表明,Strainy 能组装出准确、完整的菌株单倍型,在完整性和准确性方面优于目前基于 Nanopore 的方法,也可与基于 PacBio 的算法媲美。然后,我们使用 Strainy 组装了复杂环境元基因组的菌株单倍型,揭示了细菌物种中独特的菌株分布和突变模式。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Nature Methods
生物-生化研究方法
CiteScore
58.70
自引率
1.70%
发文量
326
审稿时长
1 months
期刊介绍:
Nature Methods is a monthly journal that focuses on publishing innovative methods and substantial enhancements to fundamental life sciences research techniques. Geared towards a diverse, interdisciplinary readership of researchers in academia and industry engaged in laboratory work, the journal offers new tools for research and emphasizes the immediate practical significance of the featured work. It publishes primary research papers and reviews recent technical and methodological advancements, with a particular interest in primary methods papers relevant to the biological and biomedical sciences. This includes methods rooted in chemistry with practical applications for studying biological problems.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


