Valid inference for machine learning-assisted genome-wide association studies
IF 31.7
1区 生物学
Q1 GENETICS & HEREDITY
引用次数: 0
Abstract
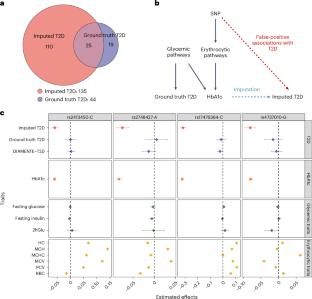
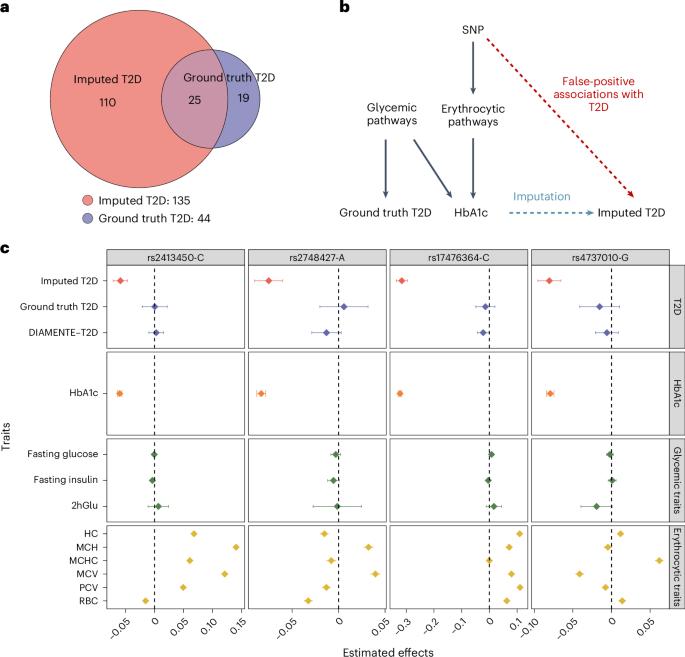
Machine learning (ML) has become increasingly popular in almost all scientific disciplines, including human genetics. Owing to challenges related to sample collection and precise phenotyping, ML-assisted genome-wide association study (GWAS), which uses sophisticated ML techniques to impute phenotypes and then performs GWAS on the imputed outcomes, have become increasingly common in complex trait genetics research. However, the validity of ML-assisted GWAS associations has not been carefully evaluated. Here, we report pervasive risks for false-positive associations in ML-assisted GWAS and introduce Post-Prediction GWAS (POP-GWAS), a statistical framework that redesigns GWAS on ML-imputed outcomes. POP-GWAS ensures valid and powerful statistical inference irrespective of imputation quality and choice of algorithm, requiring only GWAS summary statistics as input. We employed POP-GWAS to perform a GWAS of bone mineral density derived from dual-energy X-ray absorptiometry imaging at 14 skeletal sites, identifying 89 new loci and revealing skeletal site-specific genetic architecture. Our framework offers a robust analytic solution for future ML-assisted GWAS. Post-prediction genome-wide association study (POP-GWAS) is a statistical framework that uses summary statistics from labeled samples with both observed and imputed phenotypes to debias single-nucleotide polymorphism effect size estimates for unlabeled samples with imputed phenotypes only, leading to valid and powerful inference.
机器学习辅助全基因组关联研究的有效推断
机器学习(ML)在包括人类遗传学在内的几乎所有科学学科中都越来越受欢迎。由于样本收集和精确表型方面的挑战,ML 辅助全基因组关联研究(GWAS)在复杂性状遗传学研究中越来越常见,该研究使用复杂的 ML 技术来推算表型,然后对推算结果进行 GWAS。然而,ML 辅助 GWAS 关联的有效性尚未得到仔细评估。在此,我们报告了 ML 辅助 GWAS 中普遍存在的假阳性关联风险,并介绍了预测后 GWAS(POP-GWAS)--一种在 ML 估算结果上重新设计 GWAS 的统计框架。POP-GWAS 不考虑估算质量和算法选择,只需将 GWAS 摘要统计作为输入,就能确保有效且强大的统计推断。我们利用 POP-GWAS 对 14 个骨骼部位的双能 X 射线吸收仪成像得出的骨矿物质密度进行了 GWAS 分析,发现了 89 个新的基因位点,并揭示了骨骼部位特异性遗传结构。我们的框架为未来的 ML 辅助 GWAS 提供了强大的分析解决方案。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Nature genetics
生物-遗传学
CiteScore
43.00
自引率
2.60%
发文量
241
审稿时长
3 months
期刊介绍:
Nature Genetics publishes the very highest quality research in genetics. It encompasses genetic and functional genomic studies on human and plant traits and on other model organisms. Current emphasis is on the genetic basis for common and complex diseases and on the functional mechanism, architecture and evolution of gene networks, studied by experimental perturbation.
Integrative genetic topics comprise, but are not limited to:
-Genes in the pathology of human disease
-Molecular analysis of simple and complex genetic traits
-Cancer genetics
-Agricultural genomics
-Developmental genetics
-Regulatory variation in gene expression
-Strategies and technologies for extracting function from genomic data
-Pharmacological genomics
-Genome evolution
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


