Exploring Ion Polarizabilities and Their Correlation with van der Waals Radii: A Theoretical Investigation
IF 5.7
1区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
Polarizability (α) is a fundamental property which measures the tendency of the electron cloud of an atom, ion, or molecule to be distorted by electric field. Polarizability contributes to important physical properties such as molecular interactions or dielectric constants; thus, it is essential to have accurate polarizabilities in molecular simulations. However, it remains a challenge to develop polarizable force fields (FFs) for ions in computational chemistry. In particular, a comprehensive set of polarizabilities for ions has not been derived. Herein, we derived a systematic set of polarizabilities for atoms and ions across the periodic table based on high-level quantum mechanics calculations. These values have excellent agreement with experimental data. Furthermore, we examined the relationship between the obtained polarizabilities and the van der Waals (VDW) radii (RVDW) that we previously determined (J. Chem. Theory Comput., 2023, 19, 2064). Two relationships, RVDW ∝ α1/7 and RVDW ∝ α1/3, proposed in previous studies were examined in the present work. Our results indicated the former relationship, which was derived based on the quantum harmonic oscillator model, prevails for atoms and cations, but neither relationship provides a satisfactory fit for anions. This is consistent with the tight-binding nature of the electrons in atoms and cations, while it is more challenging to quantify the polarizabilities of anions because of their more dispersed electron clouds. Moreover, we compared different approaches to determine the dispersion coefficients, including the London equation, Slater–Kirkwood equation, symmetry-adapted perturbation theory (SAPT) calculations, and time-dependent density functional theory method, along with the approach based on VDW constants. Our results indicated that although different approaches predict deviated magnitudes for the dispersion coefficients, their predictions are highly correlated, implying that each of these approaches can be used to evaluate dispersion interactions after proper scaling. Finally, we have developed a parametrization strategy for the 12-6-4 model based on the obtained insights. We specifically compared the performance of the 12-6-4 model with SAPT and SobEDA analyses to model interactions involving Na+/Mg2+ and various ligands containing He, Ne, Ar, H2O, NH3, [H2PO4]−, and [HPO4]2–. Our results demonstrate that the 12-6-4 parameters effectively reproduce both the total interaction energy and the individual energy components (electrostatics, exchange-repulsion, dispersion, and induction), highlighting the physical robustness of the 12-6-4 model and the effectiveness of our parametrization approach. This study has significant implications for advancing the development of next-generation ion models and polarizable FFs.
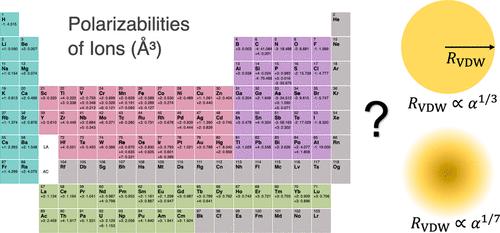
探索离子极化性及其与范德华半径的相关性:理论研究
极化性(α)是衡量原子、离子或分子的电子云在电场作用下变形趋势的基本属性。极化性对分子相互作用或介电常数等重要物理性质有影响;因此,在分子模拟中获得准确的极化性至关重要。然而,为计算化学中的离子开发可极化力场(FFs)仍然是一项挑战。特别是,目前还没有一套完整的离子极化率。在此,我们基于高水平量子力学计算,为整个元素周期表中的原子和离子推导出了一套系统的极化率。这些数值与实验数据非常吻合。此外,我们还研究了所获得的极化率与我们之前确定的范德华(VDW)半径(RVDW)之间的关系(J. Chem. Theory Comput.)本研究考察了以前研究中提出的两种关系,即 RVDW ∝ α1/7 和 RVDW ∝ α1/3。结果表明,前者是基于量子谐振子模型推导出来的,对于原子和阳离子来说更适用,但对于阴离子来说,这两种关系都不能提供令人满意的拟合效果。这与原子和阳离子中电子的紧密结合性质是一致的,而阴离子的电子云更为分散,因此量化阴离子的极化率更具挑战性。此外,我们还比较了确定弥散系数的不同方法,包括伦敦方程、斯莱特-柯克伍德方程、对称性适应扰动理论(SAPT)计算、时变密度泛函理论方法以及基于 VDW 常量的方法。我们的研究结果表明,尽管不同的方法预测的弥散系数大小存在偏差,但它们的预测结果高度相关,这意味着这些方法中的每一种都可以在适当缩放后用于评估弥散相互作用。最后,我们根据所获得的见解为 12-6-4 模型制定了参数化策略。我们特别比较了 12-6-4 模型与 SAPT 和 SobEDA 分析的性能,以模拟 Na+/Mg2+ 与含有 He、Ne、Ar、H2O、NH3、[H2PO4]- 和 [HPO4]2- 的各种配体之间的相互作用。我们的结果表明,12-6-4 参数有效地再现了总的相互作用能量和各个能量成分(静电、交换-排斥、分散和感应),突出了 12-6-4 模型的物理稳健性和我们的参数化方法的有效性。这项研究对于推动下一代离子模型和可极化 FF 的发展具有重要意义。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of Chemical Theory and Computation
化学-物理:原子、分子和化学物理
CiteScore
9.90
自引率
16.40%
发文量
568
审稿时长
1 months
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


