Adaptive Variational Quantum Computing Approaches for Green’s Functions and Nonlinear Susceptibilities
IF 5.5
1区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
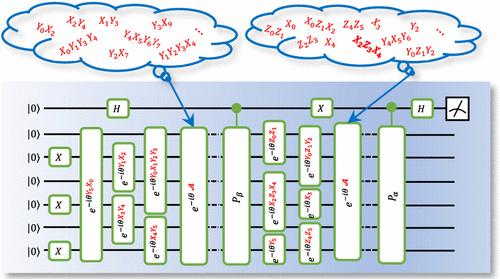
We present and benchmark quantum computing approaches for calculating real-time single-particle Green’s functions and nonlinear susceptibilities of Hamiltonian systems. The approaches leverage adaptive variational quantum algorithms for state preparation and propagation. Using automatically generated compact circuits, the dynamical evolution is performed over sufficiently long times to achieve adequate frequency resolution of the response functions. We showcase accurate Green’s function calculations using a statevector simulator on classical hardware for Fermi-Hubbard chains of 4 and 6 sites, with maximal ansatz circuit depths of 65 and 424 layers, respectively, and for the molecule LiH with a maximal ansatz circuit depth of 81 layers. Additionally, we consider an antiferromagnetic quantum spin-1 model that incorporates the Dzyaloshinskii-Moriya interaction to illustrate calculations of the third-order nonlinear susceptibilities, which can be measured in two-dimensional coherent spectroscopy experiments. These results demonstrate that real-time approaches using adaptive parametrized circuits to evaluate linear and nonlinear response functions can be feasible with near-term quantum processors.
针对格林函数和非线性易感性的自适应变分量子计算方法
我们介绍了实时计算单粒子格林函数和哈密顿系统非线性易感性的量子计算方法,并对其进行了基准测试。这些方法利用自适应变分量子算法进行状态准备和传播。利用自动生成的紧凑电路,在足够长的时间内进行动态演化,以实现响应函数的充分频率分辨率。我们展示了使用经典硬件上的状态矢量模拟器对 4 个和 6 个位点的费米-哈巴链(最大反演电路深度分别为 65 层和 424 层)以及最大反演电路深度为 81 层的分子 LiH 进行的精确格林函数计算。此外,我们还考虑了包含 Dzyaloshinskii-Moriya 相互作用的反铁磁量子自旋-1 模型,以说明三阶非线性感性的计算,这些感性可以在二维相干光谱实验中测量。这些结果表明,利用自适应参数化电路评估线性和非线性响应函数的实时方法在近期量子处理器上是可行的。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of Chemical Theory and Computation
化学-物理:原子、分子和化学物理
CiteScore
9.90
自引率
16.40%
发文量
568
审稿时长
1 months
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


