Syntheses of LSD1/HDAC Inhibitors with Demonstrated Efficacy against Colorectal Cancer: In Vitro and In Vivo Studies Including Patient-Derived Organoids
IF 6.8
1区 医学
Q1 CHEMISTRY, MEDICINAL
引用次数: 0
Abstract
Precedential evidence ascertaining the overexpression of LSD1 and HDACs in colorectal cancer spurred us to design a series of dual LSD1-HDAC inhibitors. Capitalizing on the modular nature of the three-component HDAC inhibitory model, tranylcypromine as a surface recognition motif was appended to zinc-binding motifs via diverse linkers. A compendium of hydroxamic acids was generated and evaluated for in vitro cytotoxicity against HCT-116 cells (human colorectal cancer cell lines). The most potent cell growth inhibitor 2 (GI50 = 0.495 μMm HCT-116 cells) shows promising anticancer effects by reducing colony formation and inducing cell cycle arrest in HCT-116 cells. It exhibits preferential inhibition of HDAC6, along with potent inhibition of LSD1 compared to standard inhibitors. Moreover, Compound 2 upregulates acetyl-tubulin, acetyl-histone H3, and H3K4me2, indicative of LSD1 and HDAC inhibition. In vivo, it demonstrates significant antitumor activity against colorectal cancer, better than irinotecan, and effectively inhibits growth in patient-derived CRC organoids.
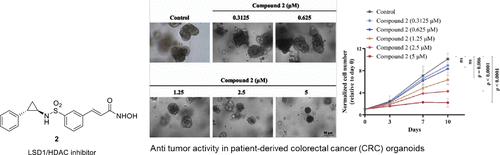
合成对结直肠癌有显著疗效的 LSD1/HDAC 抑制剂:体外和体内研究,包括患者生成的器官组织
确定 LSD1 和 HDAC 在结直肠癌中过度表达的先验证据促使我们设计出一系列 LSD1-HDAC 双重抑制剂。利用三组份 HDAC 抑制模型的模块化特性,我们通过不同的连接体将氨酰环丙胺作为表面识别基团添加到锌结合基团中。生成了羟肟酸简编,并对其对 HCT-116 细胞(人类结直肠癌细胞系)的体外细胞毒性进行了评估。最有效的细胞生长抑制剂 2(GI50 = 0.495 μMm HCT-116 细胞)通过减少 HCT-116 细胞的集落形成和诱导细胞周期停滞,显示出良好的抗癌效果。与标准抑制剂相比,它对 HDAC6 有优先抑制作用,对 LSD1 也有强效抑制作用。此外,化合物 2 还能上调乙酰基-微管蛋白、乙酰基组蛋白 H3 和 H3K4me2,这表明它抑制了 LSD1 和 HDAC。在体内,化合物 2 对结直肠癌具有显著的抗肿瘤活性,优于伊立替康,并能有效抑制源自患者的结直肠癌组织细胞的生长。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of Medicinal Chemistry
医学-医药化学
CiteScore
4.00
自引率
11.00%
发文量
804
审稿时长
1.9 months
期刊介绍:
The Journal of Medicinal Chemistry is a prestigious biweekly peer-reviewed publication that focuses on the multifaceted field of medicinal chemistry. Since its inception in 1959 as the Journal of Medicinal and Pharmaceutical Chemistry, it has evolved to become a cornerstone in the dissemination of research findings related to the design, synthesis, and development of therapeutic agents.
The Journal of Medicinal Chemistry is recognized for its significant impact in the scientific community, as evidenced by its 2022 impact factor of 7.3. This metric reflects the journal's influence and the importance of its content in shaping the future of drug discovery and development. The journal serves as a vital resource for chemists, pharmacologists, and other researchers interested in the molecular mechanisms of drug action and the optimization of therapeutic compounds.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


