Abhishek Mitra, Ruhee D’Cunha, Qiaohong Wang, Matthew R. Hermes, Yuri Alexeev, Stephen K. Gray*, Matthew Otten* and Laura Gagliardi*,
{"title":"The Localized Active Space Method with Unitary Selective Coupled Cluster","authors":"Abhishek Mitra, Ruhee D’Cunha, Qiaohong Wang, Matthew R. Hermes, Yuri Alexeev, Stephen K. Gray*, Matthew Otten* and Laura Gagliardi*, ","doi":"10.1021/acs.jctc.4c0052810.1021/acs.jctc.4c00528","DOIUrl":null,"url":null,"abstract":"<p >We introduce a hybrid quantum-classical algorithm, the localized active space unitary selective coupled cluster singles and doubles (LAS-USCCSD) method. Derived from the localized active space unitary coupled cluster (LAS-UCCSD) method, LAS-USCCSD first performs a classical LASSCF calculation, then selectively identifies the most important parameters (cluster amplitudes used to build the multireference UCC ansatz) for restoring interfragment interaction energy using this reduced set of parameters with the variational quantum eigensolver method. We benchmark LAS-USCCSD against LAS-UCCSD by calculating the total energies of (H<sub>2</sub>)<sub>2</sub>, (H<sub>2</sub>)<sub>4</sub>, and <i>trans</i>-butadiene, and the magnetic coupling constant for a bimetallic compound [Cr<sub>2</sub>(OH)<sub>3</sub>(NH<sub>3</sub>)<sub>6</sub>]<sup>3+</sup>. For these systems, we find that LAS-USCCSD reduces the number of required parameters and thus the circuit depth by at least 1 order of magnitude, an aspect which is important for the practical implementation of multireference hybrid quantum-classical algorithms like LAS-UCCSD on near-term quantum computers.</p>","PeriodicalId":5,"journal":{"name":"ACS Applied Materials & Interfaces","volume":null,"pages":null},"PeriodicalIF":8.3000,"publicationDate":"2024-09-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Applied Materials & Interfaces","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.4c00528","RegionNum":2,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MATERIALS SCIENCE, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
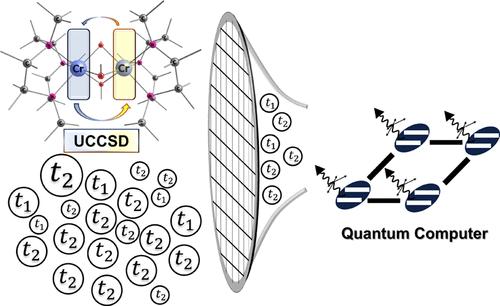
We introduce a hybrid quantum-classical algorithm, the localized active space unitary selective coupled cluster singles and doubles (LAS-USCCSD) method. Derived from the localized active space unitary coupled cluster (LAS-UCCSD) method, LAS-USCCSD first performs a classical LASSCF calculation, then selectively identifies the most important parameters (cluster amplitudes used to build the multireference UCC ansatz) for restoring interfragment interaction energy using this reduced set of parameters with the variational quantum eigensolver method. We benchmark LAS-USCCSD against LAS-UCCSD by calculating the total energies of (H2)2, (H2)4, and trans-butadiene, and the magnetic coupling constant for a bimetallic compound [Cr2(OH)3(NH3)6]3+. For these systems, we find that LAS-USCCSD reduces the number of required parameters and thus the circuit depth by at least 1 order of magnitude, an aspect which is important for the practical implementation of multireference hybrid quantum-classical algorithms like LAS-UCCSD on near-term quantum computers.
期刊介绍:
ACS Applied Materials & Interfaces is a leading interdisciplinary journal that brings together chemists, engineers, physicists, and biologists to explore the development and utilization of newly-discovered materials and interfacial processes for specific applications. Our journal has experienced remarkable growth since its establishment in 2009, both in terms of the number of articles published and the impact of the research showcased. We are proud to foster a truly global community, with the majority of published articles originating from outside the United States, reflecting the rapid growth of applied research worldwide.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


