Federated analysis of autosomal recessive coding variants in 29,745 developmental disorder patients from diverse populations
IF 31.7
1区 生物学
Q1 GENETICS & HEREDITY
引用次数: 0
Abstract
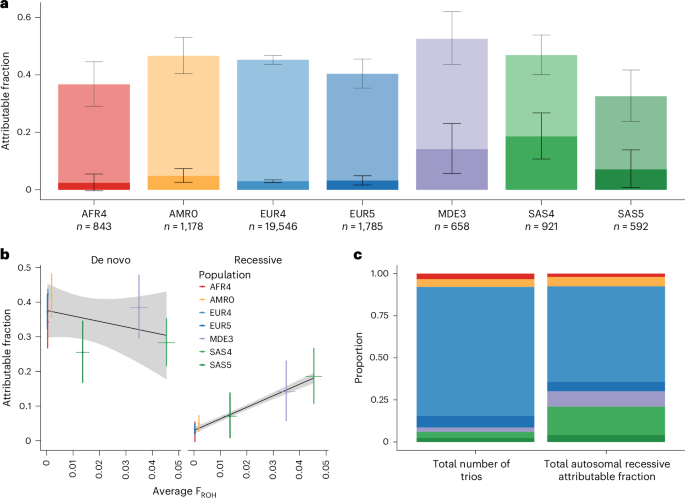
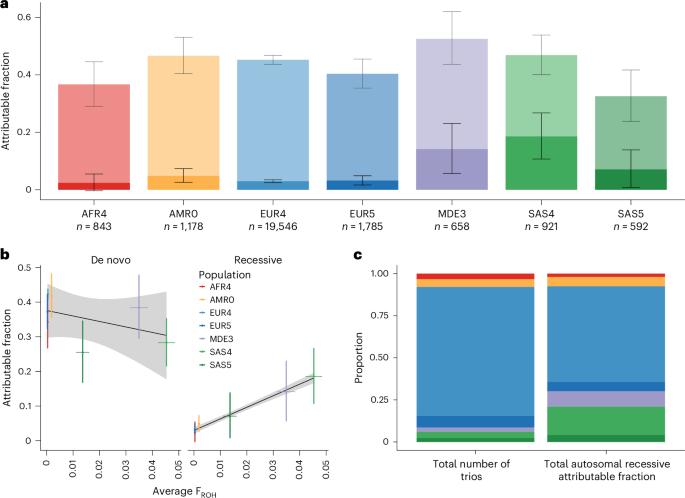
Autosomal recessive coding variants are well-known causes of rare disorders. We quantified the contribution of these variants to developmental disorders in a large, ancestrally diverse cohort comprising 29,745 trios, of whom 20.4% had genetically inferred non-European ancestries. The estimated fraction of patients attributable to exome-wide autosomal recessive coding variants ranged from ~2–19% across genetically inferred ancestry groups and was significantly correlated with average autozygosity. Established autosomal recessive developmental disorder-associated (ARDD) genes explained 84.0% of the total autosomal recessive coding burden, and 34.4% of the burden in these established genes was explained by variants not already reported as pathogenic in ClinVar. Statistical analyses identified two novel ARDD genes: KBTBD2 and ZDHHC16. This study expands our understanding of the genetic architecture of developmental disorders across diverse genetically inferred ancestry groups and suggests that improving strategies for interpreting missense variants in known ARDD genes may help diagnose more patients than discovering the remaining genes. Analysis of autosomal recessive coding variants in 29,745 trios from the DDD study and GeneDx provides insights into the genetic architecture of developmental disorders across ancestrally diverse populations.
对来自不同人群的 29 745 名发育障碍患者的常染色体隐性编码变异进行联合分析
常染色体隐性编码变异是导致罕见疾病的众所周知的原因。我们在一个由 29,745 人组成的大型祖先多样性队列中量化了这些变异对发育障碍的贡献,其中 20.4% 的人的遗传推断祖先为非欧洲人。据估计,全外显子常染色体隐性编码变异导致的患者比例在各遗传推断祖先群体中介于~2%-19%之间,并与平均自身杂合度显著相关。已确定的常染色体隐性发育障碍相关基因(ARDD)占常染色体隐性编码负担总量的 84.0%,而这些已确定基因中 34.4% 的负担是由 ClinVar 中尚未报告为致病的变异所造成的。统计分析发现了两个新的 ARDD 基因:KBTBD2 和 ZDHHC16。这项研究拓展了我们对不同遗传推断祖先群体发育障碍遗传结构的了解,并表明改进已知 ARDD 基因中错义变异的解释策略可能比发现其余基因更有助于诊断更多患者。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Nature genetics
生物-遗传学
CiteScore
43.00
自引率
2.60%
发文量
241
审稿时长
3 months
期刊介绍:
Nature Genetics publishes the very highest quality research in genetics. It encompasses genetic and functional genomic studies on human and plant traits and on other model organisms. Current emphasis is on the genetic basis for common and complex diseases and on the functional mechanism, architecture and evolution of gene networks, studied by experimental perturbation.
Integrative genetic topics comprise, but are not limited to:
-Genes in the pathology of human disease
-Molecular analysis of simple and complex genetic traits
-Cancer genetics
-Agricultural genomics
-Developmental genetics
-Regulatory variation in gene expression
-Strategies and technologies for extracting function from genomic data
-Pharmacological genomics
-Genome evolution
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


