Guang Chen , Xiaoming Chen , Ke Wang , Yaofeng Wu , Xiaolong Zhang
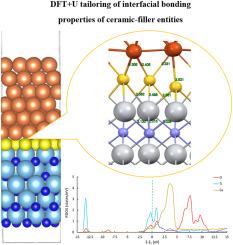
{"title":"The invisible hand of pre-adsorbates: Unveiling oxygen's role in sculpting Cu–TiN interfaces","authors":"Guang Chen , Xiaoming Chen , Ke Wang , Yaofeng Wu , Xiaolong Zhang","doi":"10.1016/j.jpcs.2024.112346","DOIUrl":null,"url":null,"abstract":"<div><p>Copper (Cu) binding to titanium nitride (TiN) surfaces is important for applications in catalysis, sensing, and electronics. However, achieving controlled and stable Cu attachment remains challenging. In this study, Density Functional Theory with Hubbard U corrections (DFT + U) is employed to investigate how pre-adsorbed oxygen (O) and sulfur (S) influence Cu attachment and the resulting interfacial properties. Contrary to initial expectations, our calculations reveal that the presence of pre-adsorbed O and S significantly weakens the Cu–TiN interface. Direct adsorption of Cu on the TiN surface, without pre-adsorbed species, results in a much higher adsorption energy (−13.94 eV), demonstrating stronger interfacial stability compared to systems with O (−2.06 eV) or S (−1.84 eV) pre-adsorption. Although pre-adsorbed O and S can modify the interface's electronic structure, the introduction of these species ultimately weakens the Cu–TiN interaction rather than enhancing it, as initially hypothesized. Analysis of the density of states (DOS) and charge transfer shows that direct Cu–TiN bonding maintains a more robust interaction, making it more suitable for applications requiring strong metal-ceramic interfaces. These findings highlight the critical role of surface chemistry in controlling the strength of Cu–TiN interfaces. The use of DFT + U calculations provides valuable insights into the bonding mechanisms and electronic changes at these interfaces, informing future design strategies for Cu–TiN systems with tailored properties for advanced technological applications.</p></div>","PeriodicalId":16811,"journal":{"name":"Journal of Physics and Chemistry of Solids","volume":"196 ","pages":"Article 112346"},"PeriodicalIF":4.3000,"publicationDate":"2024-09-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Physics and Chemistry of Solids","FirstCategoryId":"88","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0022369724004815","RegionNum":3,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
Copper (Cu) binding to titanium nitride (TiN) surfaces is important for applications in catalysis, sensing, and electronics. However, achieving controlled and stable Cu attachment remains challenging. In this study, Density Functional Theory with Hubbard U corrections (DFT + U) is employed to investigate how pre-adsorbed oxygen (O) and sulfur (S) influence Cu attachment and the resulting interfacial properties. Contrary to initial expectations, our calculations reveal that the presence of pre-adsorbed O and S significantly weakens the Cu–TiN interface. Direct adsorption of Cu on the TiN surface, without pre-adsorbed species, results in a much higher adsorption energy (−13.94 eV), demonstrating stronger interfacial stability compared to systems with O (−2.06 eV) or S (−1.84 eV) pre-adsorption. Although pre-adsorbed O and S can modify the interface's electronic structure, the introduction of these species ultimately weakens the Cu–TiN interaction rather than enhancing it, as initially hypothesized. Analysis of the density of states (DOS) and charge transfer shows that direct Cu–TiN bonding maintains a more robust interaction, making it more suitable for applications requiring strong metal-ceramic interfaces. These findings highlight the critical role of surface chemistry in controlling the strength of Cu–TiN interfaces. The use of DFT + U calculations provides valuable insights into the bonding mechanisms and electronic changes at these interfaces, informing future design strategies for Cu–TiN systems with tailored properties for advanced technological applications.
铜(Cu)与氮化钛(TiN)表面的结合对于催化、传感和电子领域的应用非常重要。然而,实现可控且稳定的铜附着仍然具有挑战性。在这项研究中,我们采用了带有哈伯德 U 修正(DFT + U)的密度泛函理论来研究预吸附的氧(O)和硫(S)如何影响铜的附着以及由此产生的界面特性。与最初的预期相反,我们的计算显示,预吸附 O 和 S 的存在会显著削弱铜-氮化钛界面。与有 O(-2.06 eV)或 S(-1.84 eV)预吸附的体系相比,没有预吸附物种的 TiN 表面直接吸附 Cu 会产生更高的吸附能(-13.94 eV),从而显示出更强的界面稳定性。虽然预吸附 O 和 S 可以改变界面的电子结构,但这些物种的引入最终会削弱而不是像最初假设的那样增强 Cu-TiN 的相互作用。对状态密度(DOS)和电荷转移的分析表明,直接的 Cu-TiN 键合能保持更强的相互作用,因此更适合需要强金属陶瓷界面的应用。这些发现凸显了表面化学在控制铜-氮化钛界面强度方面的关键作用。DFT + U 计算为了解这些界面上的成键机制和电子变化提供了宝贵的见解,为未来设计具有定制特性的 Cu-TiN 系统提供了信息,以满足先进技术应用的需要。
期刊介绍:
The Journal of Physics and Chemistry of Solids is a well-established international medium for publication of archival research in condensed matter and materials sciences. Areas of interest broadly include experimental and theoretical research on electronic, magnetic, spectroscopic and structural properties as well as the statistical mechanics and thermodynamics of materials. The focus is on gaining physical and chemical insight into the properties and potential applications of condensed matter systems.
Within the broad scope of the journal, beyond regular contributions, the editors have identified submissions in the following areas of physics and chemistry of solids to be of special current interest to the journal:
Low-dimensional systems
Exotic states of quantum electron matter including topological phases
Energy conversion and storage
Interfaces, nanoparticles and catalysts.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


