Cristian Guerra , Yeray A. Rodríguez-Núñez , Efraín Polo-Cuadrado , Adolfo Ensuncho
{"title":"Topological bonding fingerprints in photochemically substituted [2 + 2] cycloaddition","authors":"Cristian Guerra , Yeray A. Rodríguez-Núñez , Efraín Polo-Cuadrado , Adolfo Ensuncho","doi":"10.1016/j.jphotochem.2024.116038","DOIUrl":null,"url":null,"abstract":"<div><p>Using the framework of the bonding evolution theory (BET), we investigated excited-state substituted [2 + 2] cycloaddition. Our findings demonstrate that the presence of non-bonding density centers during the S<sub>1</sub> excited state results in the electronic activation of both unsubstituted and substituted ethylene. It should be noted that these electronic rearrangements imply a very high energy barrier in the ground state, where [2 + 2] cycloaddition is forbidden by the orbital symmetry rules. A crucial bonding process that leads to C<img>C bond formation in both the ground and excited states is the presence of non-bonding centers. Hence, the nature of C<img>C bond formation changes when electron-withdrawing or electron-donating groups are present in the reaction center. On the other hand, the non-polar behavior in cycloadditions is associated with low differences in electron density, whereas polar effects due to hydroxy and cyano substitutions emerge when the difference in electron density between the C<img>C bonding centers is substantial. Consequently, the topological fingerprints of the C<img>C bond creation in the photochemically induced [2 + 2] cycloadditions can be <em>cusp</em> (symmetric collapse of pairing density) if the reaction center is unsubstituted or fold if the reaction center undergoes some substitution (asymmetric collapse of pairing density).</p></div>","PeriodicalId":16782,"journal":{"name":"Journal of Photochemistry and Photobiology A-chemistry","volume":"459 ","pages":"Article 116038"},"PeriodicalIF":4.1000,"publicationDate":"2024-09-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Photochemistry and Photobiology A-chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1010603024005823","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
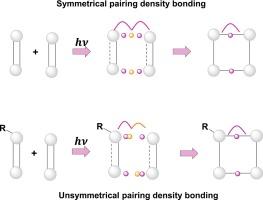
Using the framework of the bonding evolution theory (BET), we investigated excited-state substituted [2 + 2] cycloaddition. Our findings demonstrate that the presence of non-bonding density centers during the S1 excited state results in the electronic activation of both unsubstituted and substituted ethylene. It should be noted that these electronic rearrangements imply a very high energy barrier in the ground state, where [2 + 2] cycloaddition is forbidden by the orbital symmetry rules. A crucial bonding process that leads to CC bond formation in both the ground and excited states is the presence of non-bonding centers. Hence, the nature of CC bond formation changes when electron-withdrawing or electron-donating groups are present in the reaction center. On the other hand, the non-polar behavior in cycloadditions is associated with low differences in electron density, whereas polar effects due to hydroxy and cyano substitutions emerge when the difference in electron density between the CC bonding centers is substantial. Consequently, the topological fingerprints of the CC bond creation in the photochemically induced [2 + 2] cycloadditions can be cusp (symmetric collapse of pairing density) if the reaction center is unsubstituted or fold if the reaction center undergoes some substitution (asymmetric collapse of pairing density).
期刊介绍:
JPPA publishes the results of fundamental studies on all aspects of chemical phenomena induced by interactions between light and molecules/matter of all kinds.
All systems capable of being described at the molecular or integrated multimolecular level are appropriate for the journal. This includes all molecular chemical species as well as biomolecular, supramolecular, polymer and other macromolecular systems, as well as solid state photochemistry. In addition, the journal publishes studies of semiconductor and other photoactive organic and inorganic materials, photocatalysis (organic, inorganic, supramolecular and superconductor).
The scope includes condensed and gas phase photochemistry, as well as synchrotron radiation chemistry. A broad range of processes and techniques in photochemistry are covered such as light induced energy, electron and proton transfer; nonlinear photochemical behavior; mechanistic investigation of photochemical reactions and identification of the products of photochemical reactions; quantum yield determinations and measurements of rate constants for primary and secondary photochemical processes; steady-state and time-resolved emission, ultrafast spectroscopic methods, single molecule spectroscopy, time resolved X-ray diffraction, luminescence microscopy, and scattering spectroscopy applied to photochemistry. Papers in emerging and applied areas such as luminescent sensors, electroluminescence, solar energy conversion, atmospheric photochemistry, environmental remediation, and related photocatalytic chemistry are also welcome.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


