{"title":"Single-cell transcriptomic atlas reveals immune and metabolism perturbation of depression in the pathogenesis of breast cancer","authors":"Lingling Wu, Junwei Liu, Yimeng Geng, Jianwen Fang, Xingle Gao, Jianbo Lai, Minya Yao, Shaojia Lu, Weiwei Yin, Peifen Fu, Wei Chen, Shaohua Hu","doi":"10.1002/cac2.12603","DOIUrl":null,"url":null,"abstract":"<p>Epidemiological evidence indicates that major depressive disorder (MDD) may predispose the development and prognosis of breast cancer (BC) in females [<span>1</span>]. However, the mechanisms linking these phenotypes are not fully understood. Chronic stress, a hallmark of depression, has been underscored to affect anti-tumor immunity, tumor metabolic reprogramming, hormone synthesis in BC [<span>2, 3</span>], and increase tumor metastasis [<span>4</span>], but there is a lack of detailed cellular-level characterization of how MDD history affects the tumorigenesis of BC. This study explored the single-cell atlas of multiple tissues from BC patients with and without a history of MDD for characterizing the potential molecular alternations in their tumorigenesis (Figure 1A).</p><p>Paired primary tumor tissues (<i>n</i> = 10), adjacent normal tissues (<i>n</i> = 7), and peripheral blood samples (<i>n</i> = 10) were collected from a cohort of 10 BC patients, 5 of whom had a history of MDD (Supplementary Table S1). All BC patients had estrogen receptor (ER)-positive tumors and were further predicted as Luminal A (<i>n</i> = 9) and B subtypes (<i>n</i> = 1) (Supplementary Table S2). Further details on patient recruitment, sample handling, and single-cell data analysis are provided in Supplementary Methods. In total, we obtained 224,557 single cells and further annotated them into major cell subsets based on lineage markers and copy number variations [<span>5</span>] (Figure 1B, Supplementary Figure S1A-B). Aneuploid cells in primary tumor tissues were obtained (Supplementary Table S3) to characterize their phenotypic differences in BC patients with MDD history (BC-MDD) or not (BC-Ctrl). The Uniform Manifold Approximation and Projection (UMAP) of unintegrated aneuploid cells revealed intrinsic differences across individual patients (Supplementary Figure S1C). Downstream functional profiling analysis identified distinct immune response pathways in BC-MDD and BC-Ctrl groups and enrichment of the oxidative phosphorylation (OXPHOS) pathway in BC-Ctrl tumors (Supplementary Figure S1D-E). Cellular Gene Set Variation Analysis (GSVA) [<span>6</span>] confirmed the distinct metabolic phenotypes between BC-MDD and BC-Ctrl groups (Figure 1C). Additionally, utilizing predefined gene module (GM) signatures of BC tumor cells [<span>7</span>], we observed specific restraint of GM4 and GM6 in BC-MDD (Supplementary Figure S1F-G). GM6 encompasses various antigen presentation genes, aligning with the observed downregulation of major histocompatibility complex class I (MHC-I) class genes in BC-MDD tumor cells (Supplementary Figure S1H).</p><p>Upon re-clustering 12,371 normal epithelial cells within primary breast tumor and adjacent normal tissues from the 10 patients, we identified 9 cell clusters, including luminal hormone-responsive (LumHR), luminal secretory (LumSec), and myoepithelial cells [<span>8</span>] (Supplementary Figure S2A-B). Distribution analysis suggested the potential enrichment of LumSec-2 and -3 clusters in primary tumor tissues of BC-Ctrl patients (Supplementary Figure S2C-E). Subsequently, we investigated the potential roles of these BC-Ctrl-enriched tumor epithelial cells in tumorigenesis and prognosis. We utilized the top 50 differentially expressed genes (DEGs) of these cell clusters to evaluate their prognostic roles using The Cancer Genome Atlas-Breast Invasive Carcinoma (TCGA-BRCA) datasets [<span>9</span>]. Notably, we revealed that the gene signatures of these cells could only predict the prognosis of ER<sup>+</sup> but not ER<sup>−</sup> patients (Figure 1D, Supplementary Figure S2F). Despite the high expression of basal and human epidermal growth factor receptor 2 (HER2) breast cancer phenotypes, the prognostic roles of these cells may be related to the downregulation of prostate-derived ETS factor (PDEF/SPDEF) [<span>10</span>] (Supplementary Figure S2G-H).</p><p>We then integrated and characterized the functional profiling of 25,076 stromal cells within primary breast tumor and adjacent normal tissues, encompassing 7 endothelial cell, 10 fibroblast, and 5 pericyte clusters (Supplementary Figure S3A-E). Frequency distribution analyses identified the tumor-specific cell cluster enrichment but not across patient groups in both tissue sites (Supplementary Figure S3F-H). We then characterized the potential MDD-specific functional changes within stromal cells, and noted consistent functional changes across all major stromal subtypes. We identified 70 upregulated genes shared by all stromal subtypes in BC-MDD patients, with pathway analysis suggested their involvement in metabolic regulation and cellular stress response pathways, consistent with trends observed in individual patients (Figure 1E, Supplementary Figure S3I). Specifically, we found the potential up-regulation of solute carrier family 38 member 2 (<i>SLC38A2</i>) and heat shock protein family H member 1 (<i>HSPH1</i>) in primary breast tumor and adjacent normal tissues from BC-MDD patients (Supplementary Figure S3J), highlighting the potential roles of amino acid transport and stress responses in MDD-affected BC patients.</p><p>To characterize the T cell immune response alterations in BC-MDD patients, we re-integrated 91,615 annotated T/natural killer (NK) cells from primary breast tumors, adjacent normal tissues, and peripheral blood samples. This analysis identified 15 distinct cell clusters (Supplementary Figure S4A-B). Notably, C-X-C motif chemokine ligand 13 (<i>CXCL13</i>)<sup>+</sup> CD4<sup>+</sup> T cells (CD4-Tem-2) and regulatory T cells were enriched in primary tumors, NK and γδT cells were enriched in peripheral blood (Supplementary Figure S4C). However, the frequency comparisons of T/NK cell clusters across patient groups revealed non-significant differences across all tissues (Supplementary Figure S4D). We then profiled the functional alternations in CD8<sup>+</sup> T cells in primary tumors with MDD history, which proposed the enrichment of interferon-related genes and cytotoxic genes in BC-Ctrl tumor tissues, validated with individual patients (Supplementary Figure S4E-F). This finding is consistent with the diminished activation of immune response and cytokine response pathways in CD8 T cells in the BC-MDD group (Figure 1F).</p><p>We conducted a parallel analysis with 27,563 myeloid cells and identified 16 cell clusters within monocytes, macrophages, and dendritic cells (DCs) (Supplementary Figure S5A-B). Among those, the Macro-m1-1 cell cluster, characterized by high expression of chemokine genes, predominated in primary tumor tissues, alongside the G protein-coupled receptor 183 (<i>GPR183</i>)-positive cDC-2 cells (Supplementary Figure S5C). Conversely, we observed no significant distribution variations in myeloid cells across all tissues among patient groups (Supplementary Figure S5D). Exploring the functional modifications of macrophages in primary tumor tissues, we uncovered enrichment of fatty acid-binding genes in the BC-MDD group and interferon-related genes in the BC-Ctrl group, which were validated across individual patients (Supplementary Figure S5E-F). Similar to CD8<sup>+</sup> T cells, we revealed enrichment of stress response and metabolic-related pathways but with the impairment of innate immune response pathways in the BC-MDD group (Figure 1F).</p><p>We next compared the ligand-receptor interactions within primary tumor tissues, and observed differences in interaction number among major cell types between BC-Ctrl and BC-MDD groups (Figure 1G). In the BC-MDD group, the signal strength for interactions showed a significant reduction in epithelial cells, followed by fibroblasts and tumor cells, while signals related to T/NK cells exhibited a slight enhancement (Figure 1H). We then focused on the interaction changes within tumor cells, epithelial cells, and T/NK cells. Within T/NK cells, the C-X-C chemokine receptor 4 and ligand 12 (<i>CXCR4</i>-<i>CXCL12</i>) axis and macrophage migration inhibitory factor (MIF) signal were upregulated in the BC-MDD group (Supplementary Figure S6A-B). Meanwhile, we identified the enrichment in signals of midkine (MDK), laminin, and fibronectin (FN1) in the BC-Ctrl group (Supplementary Figure S6C-D). These results suggest altered breast cancer tumor signal interactions associated with a history of MDD.</p><p>In summary, our study offers preliminary insights regarding the impacts of MDD history in BC patients. We suggest a potential association between MDD history and a poorer prognosis in BC, characterized by a decrease in specific epithelial cells and impaired immune regulation signals within primary tumors. Furthermore, we propose potential alterations in the metabolic status of BC tumor cells in patients with MDD. Given the constraints of sample size and limited follow-up duration, larger patient cohorts, longitudinal data, and additional validation experiments are necessary to fully understand the complex interplay between MDD history and BC.</p><p>The authors declare that they have no competing interests.</p><p>This work was supported by funding from the Zhejiang Provincial Key Research and Development Program (No. 2021C03107 to S.H.), the Leading Talent of Scientific and Technological Innovation - “Ten Thousand Talents Program” of Zhejiang Province (No. 2021R52016 to S.H.), the National Natural Science Foundation (No. 82172770 to P.F.), and the Fundamental Research Funds for the Central Universities (226-2022-00193, 226-2022-00002).</p><p>The study was reviewed and approved by the First Affiliated Hospital, Zhejiang University School of Medicine (Approval number: IIT20220235B) and conducted in conformity with the International Ethical Guidelines for Research Involving Human Subjects as stated in the Helsinki Declaration. Informed written consent was obtained from all patients in this study.</p>","PeriodicalId":9495,"journal":{"name":"Cancer Communications","volume":"44 11","pages":"1311-1315"},"PeriodicalIF":20.1000,"publicationDate":"2024-09-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/cac2.12603","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cancer Communications","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/cac2.12603","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"ONCOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
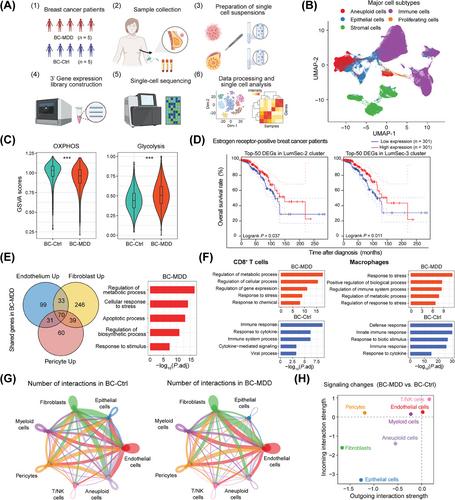
Epidemiological evidence indicates that major depressive disorder (MDD) may predispose the development and prognosis of breast cancer (BC) in females [1]. However, the mechanisms linking these phenotypes are not fully understood. Chronic stress, a hallmark of depression, has been underscored to affect anti-tumor immunity, tumor metabolic reprogramming, hormone synthesis in BC [2, 3], and increase tumor metastasis [4], but there is a lack of detailed cellular-level characterization of how MDD history affects the tumorigenesis of BC. This study explored the single-cell atlas of multiple tissues from BC patients with and without a history of MDD for characterizing the potential molecular alternations in their tumorigenesis (Figure 1A).
Paired primary tumor tissues (n = 10), adjacent normal tissues (n = 7), and peripheral blood samples (n = 10) were collected from a cohort of 10 BC patients, 5 of whom had a history of MDD (Supplementary Table S1). All BC patients had estrogen receptor (ER)-positive tumors and were further predicted as Luminal A (n = 9) and B subtypes (n = 1) (Supplementary Table S2). Further details on patient recruitment, sample handling, and single-cell data analysis are provided in Supplementary Methods. In total, we obtained 224,557 single cells and further annotated them into major cell subsets based on lineage markers and copy number variations [5] (Figure 1B, Supplementary Figure S1A-B). Aneuploid cells in primary tumor tissues were obtained (Supplementary Table S3) to characterize their phenotypic differences in BC patients with MDD history (BC-MDD) or not (BC-Ctrl). The Uniform Manifold Approximation and Projection (UMAP) of unintegrated aneuploid cells revealed intrinsic differences across individual patients (Supplementary Figure S1C). Downstream functional profiling analysis identified distinct immune response pathways in BC-MDD and BC-Ctrl groups and enrichment of the oxidative phosphorylation (OXPHOS) pathway in BC-Ctrl tumors (Supplementary Figure S1D-E). Cellular Gene Set Variation Analysis (GSVA) [6] confirmed the distinct metabolic phenotypes between BC-MDD and BC-Ctrl groups (Figure 1C). Additionally, utilizing predefined gene module (GM) signatures of BC tumor cells [7], we observed specific restraint of GM4 and GM6 in BC-MDD (Supplementary Figure S1F-G). GM6 encompasses various antigen presentation genes, aligning with the observed downregulation of major histocompatibility complex class I (MHC-I) class genes in BC-MDD tumor cells (Supplementary Figure S1H).
Upon re-clustering 12,371 normal epithelial cells within primary breast tumor and adjacent normal tissues from the 10 patients, we identified 9 cell clusters, including luminal hormone-responsive (LumHR), luminal secretory (LumSec), and myoepithelial cells [8] (Supplementary Figure S2A-B). Distribution analysis suggested the potential enrichment of LumSec-2 and -3 clusters in primary tumor tissues of BC-Ctrl patients (Supplementary Figure S2C-E). Subsequently, we investigated the potential roles of these BC-Ctrl-enriched tumor epithelial cells in tumorigenesis and prognosis. We utilized the top 50 differentially expressed genes (DEGs) of these cell clusters to evaluate their prognostic roles using The Cancer Genome Atlas-Breast Invasive Carcinoma (TCGA-BRCA) datasets [9]. Notably, we revealed that the gene signatures of these cells could only predict the prognosis of ER+ but not ER− patients (Figure 1D, Supplementary Figure S2F). Despite the high expression of basal and human epidermal growth factor receptor 2 (HER2) breast cancer phenotypes, the prognostic roles of these cells may be related to the downregulation of prostate-derived ETS factor (PDEF/SPDEF) [10] (Supplementary Figure S2G-H).
We then integrated and characterized the functional profiling of 25,076 stromal cells within primary breast tumor and adjacent normal tissues, encompassing 7 endothelial cell, 10 fibroblast, and 5 pericyte clusters (Supplementary Figure S3A-E). Frequency distribution analyses identified the tumor-specific cell cluster enrichment but not across patient groups in both tissue sites (Supplementary Figure S3F-H). We then characterized the potential MDD-specific functional changes within stromal cells, and noted consistent functional changes across all major stromal subtypes. We identified 70 upregulated genes shared by all stromal subtypes in BC-MDD patients, with pathway analysis suggested their involvement in metabolic regulation and cellular stress response pathways, consistent with trends observed in individual patients (Figure 1E, Supplementary Figure S3I). Specifically, we found the potential up-regulation of solute carrier family 38 member 2 (SLC38A2) and heat shock protein family H member 1 (HSPH1) in primary breast tumor and adjacent normal tissues from BC-MDD patients (Supplementary Figure S3J), highlighting the potential roles of amino acid transport and stress responses in MDD-affected BC patients.
To characterize the T cell immune response alterations in BC-MDD patients, we re-integrated 91,615 annotated T/natural killer (NK) cells from primary breast tumors, adjacent normal tissues, and peripheral blood samples. This analysis identified 15 distinct cell clusters (Supplementary Figure S4A-B). Notably, C-X-C motif chemokine ligand 13 (CXCL13)+ CD4+ T cells (CD4-Tem-2) and regulatory T cells were enriched in primary tumors, NK and γδT cells were enriched in peripheral blood (Supplementary Figure S4C). However, the frequency comparisons of T/NK cell clusters across patient groups revealed non-significant differences across all tissues (Supplementary Figure S4D). We then profiled the functional alternations in CD8+ T cells in primary tumors with MDD history, which proposed the enrichment of interferon-related genes and cytotoxic genes in BC-Ctrl tumor tissues, validated with individual patients (Supplementary Figure S4E-F). This finding is consistent with the diminished activation of immune response and cytokine response pathways in CD8 T cells in the BC-MDD group (Figure 1F).
We conducted a parallel analysis with 27,563 myeloid cells and identified 16 cell clusters within monocytes, macrophages, and dendritic cells (DCs) (Supplementary Figure S5A-B). Among those, the Macro-m1-1 cell cluster, characterized by high expression of chemokine genes, predominated in primary tumor tissues, alongside the G protein-coupled receptor 183 (GPR183)-positive cDC-2 cells (Supplementary Figure S5C). Conversely, we observed no significant distribution variations in myeloid cells across all tissues among patient groups (Supplementary Figure S5D). Exploring the functional modifications of macrophages in primary tumor tissues, we uncovered enrichment of fatty acid-binding genes in the BC-MDD group and interferon-related genes in the BC-Ctrl group, which were validated across individual patients (Supplementary Figure S5E-F). Similar to CD8+ T cells, we revealed enrichment of stress response and metabolic-related pathways but with the impairment of innate immune response pathways in the BC-MDD group (Figure 1F).
We next compared the ligand-receptor interactions within primary tumor tissues, and observed differences in interaction number among major cell types between BC-Ctrl and BC-MDD groups (Figure 1G). In the BC-MDD group, the signal strength for interactions showed a significant reduction in epithelial cells, followed by fibroblasts and tumor cells, while signals related to T/NK cells exhibited a slight enhancement (Figure 1H). We then focused on the interaction changes within tumor cells, epithelial cells, and T/NK cells. Within T/NK cells, the C-X-C chemokine receptor 4 and ligand 12 (CXCR4-CXCL12) axis and macrophage migration inhibitory factor (MIF) signal were upregulated in the BC-MDD group (Supplementary Figure S6A-B). Meanwhile, we identified the enrichment in signals of midkine (MDK), laminin, and fibronectin (FN1) in the BC-Ctrl group (Supplementary Figure S6C-D). These results suggest altered breast cancer tumor signal interactions associated with a history of MDD.
In summary, our study offers preliminary insights regarding the impacts of MDD history in BC patients. We suggest a potential association between MDD history and a poorer prognosis in BC, characterized by a decrease in specific epithelial cells and impaired immune regulation signals within primary tumors. Furthermore, we propose potential alterations in the metabolic status of BC tumor cells in patients with MDD. Given the constraints of sample size and limited follow-up duration, larger patient cohorts, longitudinal data, and additional validation experiments are necessary to fully understand the complex interplay between MDD history and BC.
The authors declare that they have no competing interests.
This work was supported by funding from the Zhejiang Provincial Key Research and Development Program (No. 2021C03107 to S.H.), the Leading Talent of Scientific and Technological Innovation - “Ten Thousand Talents Program” of Zhejiang Province (No. 2021R52016 to S.H.), the National Natural Science Foundation (No. 82172770 to P.F.), and the Fundamental Research Funds for the Central Universities (226-2022-00193, 226-2022-00002).
The study was reviewed and approved by the First Affiliated Hospital, Zhejiang University School of Medicine (Approval number: IIT20220235B) and conducted in conformity with the International Ethical Guidelines for Research Involving Human Subjects as stated in the Helsinki Declaration. Informed written consent was obtained from all patients in this study.
期刊介绍:
Cancer Communications is an open access, peer-reviewed online journal that encompasses basic, clinical, and translational cancer research. The journal welcomes submissions concerning clinical trials, epidemiology, molecular and cellular biology, and genetics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


