The Effect of Silicon Substitution by Boron for the α-Nb5Si3: INSIGHTS into the Constitutive Properties of Nb5Si2B Through Theory and Experimental Approach
Michał Falkowski, Jakub Kaczkowski, Grażyna Chełkowska, Andrzej Kowalczyk
{"title":"The Effect of Silicon Substitution by Boron for the α-Nb5Si3: INSIGHTS into the Constitutive Properties of Nb5Si2B Through Theory and Experimental Approach","authors":"Michał Falkowski, Jakub Kaczkowski, Grażyna Chełkowska, Andrzej Kowalczyk","doi":"10.1007/s11661-024-07583-6","DOIUrl":null,"url":null,"abstract":"<p>We investigated the structural, elastic, vibrational, and electronic properties of the Nb<sub>5</sub>Si<sub>2</sub>B compound combining density functional theory (DFT) calculations and experimental methods. We compared our results with the parent compound Nb<sub>5</sub>Si<sub>3</sub> with two non-equivalent Si sites namely Si(4a) and Si(8h). The analysis of elastic constants and phonon spectra indicate that Nb<sub>5</sub>Si<sub>2</sub>B is respectively mechanically and dynamically stable. Based on the phonon calculation we evaluate the theoretical constant volume lattice specific heat (<i>C</i><sub>V</sub>) for different site occupancies and compare it with experimental specific heat (<i>C</i><sub>p</sub>) measurements. We found an excellent agreement between theoretical and experimental results for Nb<sub>5</sub>Si<sub>2</sub>B with the B at the Si(8h) site, which agrees with the calculated formation energy. In addition, we also performed DFT calculations aimed at showing and comparing the total DOS near the Fermi level (<i>E</i><sub>F</sub>) for Nb<sub>5</sub>Si<sub>3</sub> and Nb<sub>5</sub>Si<sub>2</sub>B. The XPS valence band (VB) of the Nb<sub>5</sub>Si<sub>2</sub>B is largely dominated by two characteristic peaks at − 8.7 and − 1.7 eV, respectively. Based on DFT calculations, it follows that the main sharp peak at − 1.7 eV comes as a contribution from Nb 4d states, while the smaller and broader one located at − 8.7 eV results mainly from Si 3s states weakly hybridized with Nb 4d states. In this connection, the majority contribution in the binding energy range from − 12 eV to the <i>E</i><sub>F</sub> comes from Nb 4d states, while the contribution from Si and B atoms is very small in this region. The core levels of Nb 3d, Si 2s, 2p, and B 1s were also identified using the XPS technique.</p>","PeriodicalId":18504,"journal":{"name":"Metallurgical and Materials Transactions A","volume":"567 1","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2024-09-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Metallurgical and Materials Transactions A","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1007/s11661-024-07583-6","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
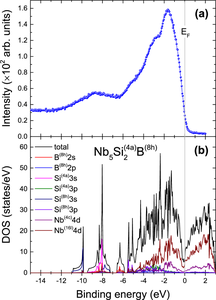
We investigated the structural, elastic, vibrational, and electronic properties of the Nb5Si2B compound combining density functional theory (DFT) calculations and experimental methods. We compared our results with the parent compound Nb5Si3 with two non-equivalent Si sites namely Si(4a) and Si(8h). The analysis of elastic constants and phonon spectra indicate that Nb5Si2B is respectively mechanically and dynamically stable. Based on the phonon calculation we evaluate the theoretical constant volume lattice specific heat (CV) for different site occupancies and compare it with experimental specific heat (Cp) measurements. We found an excellent agreement between theoretical and experimental results for Nb5Si2B with the B at the Si(8h) site, which agrees with the calculated formation energy. In addition, we also performed DFT calculations aimed at showing and comparing the total DOS near the Fermi level (EF) for Nb5Si3 and Nb5Si2B. The XPS valence band (VB) of the Nb5Si2B is largely dominated by two characteristic peaks at − 8.7 and − 1.7 eV, respectively. Based on DFT calculations, it follows that the main sharp peak at − 1.7 eV comes as a contribution from Nb 4d states, while the smaller and broader one located at − 8.7 eV results mainly from Si 3s states weakly hybridized with Nb 4d states. In this connection, the majority contribution in the binding energy range from − 12 eV to the EF comes from Nb 4d states, while the contribution from Si and B atoms is very small in this region. The core levels of Nb 3d, Si 2s, 2p, and B 1s were also identified using the XPS technique.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


