Accelerated Computer-Aided Screening of Optical Materials: Investigating the Potential of Δ-SCF Methods to Predict Emission Maxima of Large Dye Molecules
IF 2.7
2区 化学
Q3 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
Accurate simulation of electronic excited states of large chromophores is often difficult due to the computationally expensive nature of existing methods. Common approximations such as fragmentation methods that are routinely applied to ground-state calculations of large molecules are not easily applicable to excited states due to the delocalized nature of electronic excitations in most practical chromophores. Thus, special techniques specific to excited states are needed. Δ-SCF methods are one such approximation that treats excited states in a manner analogous to that for ground-state calculations, accelerating the simulation of excited states. In this work, we employed the popular initial maximum overlap method (IMOM) to avoid the variational collapse of the electronic excited state orbitals to the ground state. We demonstrate that it is possible to obtain emission energies from the first singlet (S1) excited state of many thousands of dye molecules without any external intervention. Spin correction was found to be necessary to obtain accurate excitation and emission energies. Using thousands of dye-like chromophores and various solvents (12,318 combinations), we show that the spin-corrected initial maximum overlap method accurately predicts emission maxima with a mean absolute error of only 0.27 eV. We further improved the predictive accuracy using linear fit-based corrections from individual dye classes to achieve an impressive performance of 0.17 eV. Additionally, we demonstrate that IMOM spin density can be used to identify the dye class of chromophores, enabling improved prediction accuracy for complex dye molecules, such as dyads (chromophores containing moieties from two different dye classes). Finally, the convergence behavior of IMOM excited state SCF calculations is analyzed briefly to identify the chemical space, where IMOM is more likely to fail.
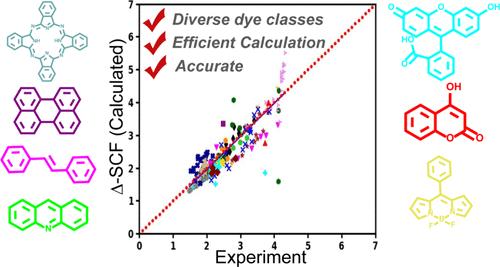
加速计算机辅助筛选光学材料:研究 Δ-SCF 方法预测大染料分子发射最大值的潜力
由于现有方法的计算成本高昂,精确模拟大型发色团的电子激发态往往十分困难。常规用于大分子基态计算的常见近似方法(如碎裂法),由于大多数实用发色团的电子激发具有非局域性,因此不易应用于激发态。因此,需要专门针对激发态的特殊技术。Δ-SCF 方法就是这样一种近似方法,它以类似于基态计算的方式处理激发态,从而加速了激发态的模拟。在这项工作中,我们采用了流行的初始最大重叠法(IMOM),以避免电子激发态轨道坍缩到基态。我们证明,在没有任何外部干预的情况下,可以获得成千上万染料分子的第一个单子激发态(S1)的发射能量。我们发现,要获得准确的激发和发射能量,必须进行自旋校正。通过使用数千种染料类发色团和各种溶剂(12,318 种组合),我们发现自旋校正初始最大重叠方法可以准确预测发射最大值,平均绝对误差仅为 0.27 eV。我们利用基于线性拟合的单类染料修正进一步提高了预测精度,达到了令人印象深刻的 0.17 eV。此外,我们还证明了 IMOM 自旋密度可用于识别发色团的染料类别,从而提高复杂染料分子的预测精度,例如二元染料(包含两个不同染料类别分子的发色团)。最后,简要分析了 IMOM 激发态 SCF 计算的收敛行为,以确定 IMOM 更容易失败的化学空间。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

The Journal of Physical Chemistry A
化学-物理:原子、分子和化学物理
CiteScore
5.20
自引率
10.30%
发文量
922
审稿时长
1.3 months
期刊介绍:
The Journal of Physical Chemistry A is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


