Excited-State Forces with the Gaussian and Augmented Plane Wave Method for the Tamm–Dancoff Approximation of Time-Dependent Density Functional Theory
IF 5.7
1区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
Augmented plane wave methods enable an efficient description of atom-centered or localized features of the electronic density, circumventing high energy cutoffs and thus prohibitive computational costs of pure plane wave formulations. To complement existing implementations for ground-state properties and excitation energies, we present the extension of the Gaussian and augmented plane wave method to excited-state nuclear gradients within the Tamm–Dancoff approximation of time-dependent density functional theory and its implementation in the CP2K program package. Benchmarks for a test set of 35 small molecules demonstrate that maximum errors in the nuclear forces for excited states of singlet and triplet spin multiplicity are smaller than 0.1 eV/Å. The method is furthermore applied to the calculation of the zero-phonon line of defective hexagonal boron nitride. This spectral feature is reproduced with an error of 0.6 eV in comparison to GW–Bethe–Salpeter reference computations and 0.4 eV in comparison to experimental measurements. Accuracy assessments and applications thus demonstrate the potential use of the outlined developments for large-scale applications on excited-state properties of extended systems.
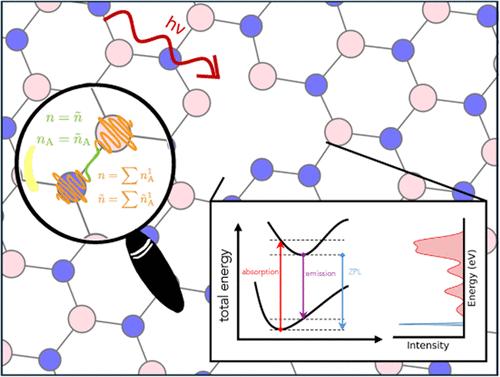
用高斯和增强平面波方法计算时变密度泛函理论的激发态作用力
增强平面波方法能够有效描述以原子为中心或局部的电子密度特征,避开了纯平面波公式的高能量截止点和令人望而却步的计算成本。为了补充现有的基态性质和激发能量的实现方法,我们介绍了高斯和增强平面波方法在时变密度泛函理论的塔姆-丹可夫近似中对激发态核梯度的扩展及其在 CP2K 程序包中的实现。对 35 个小分子测试集的基准测试表明,单重和三重自旋多重性激发态核力的最大误差小于 0.1 eV/Å。该方法还被进一步应用于计算有缺陷的六方氮化硼的零声子线。与 GW-Bethe-Salpeter 参考计算结果相比,该光谱特征的再现误差为 0.6 eV;与实验测量结果相比,误差为 0.4 eV。因此,精确度评估和应用证明了所概述的开发成果在扩展系统激发态特性的大规模应用中的潜在用途。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of Chemical Theory and Computation
化学-物理:原子、分子和化学物理
CiteScore
9.90
自引率
16.40%
发文量
568
审稿时长
1 months
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


