Dragan Jevremovic, Min Shi, Pedro Horna, Gregory E. Otteson, Michael M. Timm, Shannon A. Bennett, Linda B. Baughn, Patricia T. Greipp, Wilson I. Gonsalves, Prashant Kapoor, Morie A. Gertz, Moritz Binder, Francis K. Buadi, Angela Dispenzieri, Taxiarchis Kourelis, Eli Muchtar, Jiehao Zhou, S. Vincent Rajkumar, Shaji K. Kumar, Horatiu Olteanu
{"title":"FDA IDE validation of multiple myeloma MRD test by flow cytometry","authors":"Dragan Jevremovic, Min Shi, Pedro Horna, Gregory E. Otteson, Michael M. Timm, Shannon A. Bennett, Linda B. Baughn, Patricia T. Greipp, Wilson I. Gonsalves, Prashant Kapoor, Morie A. Gertz, Moritz Binder, Francis K. Buadi, Angela Dispenzieri, Taxiarchis Kourelis, Eli Muchtar, Jiehao Zhou, S. Vincent Rajkumar, Shaji K. Kumar, Horatiu Olteanu","doi":"10.1002/ajh.27484","DOIUrl":null,"url":null,"abstract":"<p>Two recent decisions by the Food and Drug Administration will likely significantly impact testing for multiple myeloma (MM) minimal residual disease (MRD). First, on April 12, 2024, the FDA's Oncologic Drugs Advisory Committee (ODAC) voted to approve the use of MRD as an end point for accelerated approval of new treatments for patients with MM.<span><sup>1</sup></span> This was a result of near 10 years effort by multiple institutions, professional societies, and patient's advocacy groups,<span><sup>2-6</sup></span> and reflects the current state of MM treatment in which new therapeutic options have dramatically improved progression-free and overall survival (PFS and OS),<span><sup>7</sup></span> making them impractical as the only clinical trial endpoints. Second, after many years of deliberations, FDA announced on April 29, 2024 that it will start overseeing laboratory developed tests (LDTs).<span><sup>8</sup></span> LDTs are in vitro diagnostic products (IVDs) intended for clinical use; they are designed and validated for use within individual laboratories certified for performing high complexity testing under the Clinical Laboratory Improvement Amendments of 1988 (CLIA). The FDA's decision is currently being challenged by the American Clinical Laboratory Association.</p><p>The most commonly used assays for MRD detection in plasma cell neoplasms are next-generation sequencing (NGS) of the rearranged variable immunoglobulin heavy chain<span><sup>9, 10</sup></span> and high sensitivity flow cytometry immunophenotyping (next-generation flow, NGF).<span><sup>11-14</sup></span> Each type of assay has its advantages and disadvantages. For example, NGS requires knowledge of the diagnostic specimen for sequence comparison, but that knowledge enables higher sensitivity (10<sup>−6</sup>). NGF, as developed by the Euroflow/Spanish flow cytometry experts and commercialized by Cytognos requires no prior diagnostic specimen and can provide information regarding quality of the specimen (hemodilution); the sensitivity of the NGF is between 10<sup>−5</sup> and 2 × 10<sup>−6</sup>, depending on the number of cells (events) collected. In 2016, the International Myeloma Working Group established the minimum sensitivity of 10<sup>−5</sup> for MM MRD testing.<span><sup>4</sup></span></p><p>Currently the only FDA-approved test for MM MRD is NGS-based clonoSEQ® by Adaptive Biotechnologies.<span><sup>9</sup></span> For the NGF method to be FDA-approved, it will require a comprehensive package submission to the FDA, either as Premarket Approval (PMA) application or a Premarket Notification 510(k). However, FDA has a category of investigational device exemption (IDE) which allows a test to be used in a clinical study to collect safety and effectiveness data. There is limited data available regarding the requirements for FDA IDE approval of a flow cytometry assay for MRD testing.</p><p>In our laboratory, we adopted and validated the NGF method as an LDT, and have, since 2017, tested over 13 000 unique specimens (unpublished data). Our test validation protocol had been certified by CLIA and New York State Department of Health. However, when we submitted our validation documents as a part of the application for the FDA IDE for the BIQSFP-Funded Study EAA171, FDA requested additional validation experiments due to the fact that the MRD result was going to be used for MM patient stratification. After several meetings to precisely define additional requirements, we agreed with the FDA on the plan of action. Here we summarize additional experiments required by the FDA in order to approve IDE for the use of NGF in a clinical trial.</p><p><i>Analyte, specimen, and/or matrix stability</i>: Additional four bone marrow samples were stored in shipping containers at ambient temperature and tested after 24, 48, 72, and 96 h. Results from the accepted time points had to be qualitatively the same (MRD-positive or negative). The last acceptable time point was defined as the one in which the coefficient of variation (CV) ≤25%. The data showed that under ambient shipping condition, MRD detection can be confidently assessed up to 96 h post-draw (Table S1).</p><p><i>Precision/reproducibility</i>: Additional three MM samples were spiked into normal bone marrow samples, for calculated tumor burden between 5 × 10<sup>−6</sup> and 5 × 10<sup>−5</sup>. A total of eight replicates were performed for each sample. CVs were calculated for comparisons of intra-assay, inter-assay, operator-to-operator, and instrument-to-instrument precision. Each replicate had to be qualitatively the same, with all the CVs ≤25%. Standardized machine settings and standardized gating strategy showed a highly precise assay near the assay's limit of detection (LOD) (Tables S2a and S2b).</p><p><i>Accuracy</i>: Twenty-one MM samples were used to compare MRD results by NGF with NGS-based clonoSEQ® assay, using split sample procedure. Three samples were tested with two different concentrations of spiked abnormal cells. Additional 13 diagnostic specimens were also a part of this comparison. The results showed 100% concordance of NGF with clonoSEQ® NGS testing for MRD ≥10<sup>−5</sup>. In addition, 10 out of 10 NGF-positive cases with the sensitivity between 2 × 10<sup>−6</sup> and 10<sup>−5</sup> were confirmed positive by NGS. Four NGF-negative cases showed very low level of positivity by NGS (3–4 × 10<sup>−6</sup>) (Figure 1 and Table S3). Overall Pearson correlation coefficient was 0.83.</p><p><i>Analytical sensitivity/limit of detection</i>: Four MM bone marrow samples were spiked into normal bone marrow to achieve calculated tumor burden of 10<sup>−3</sup> (baseline), 10<sup>−4</sup>, 10<sup>−5</sup>, and 5 × 10<sup>−6</sup> (half-log below the assay LOD), in triplicates, with expected CVs ≤25%. Two samples showed precise MRD-positive LOD down to 2 × 10<sup>−6</sup>, and the other two down to 10<sup>−5</sup> (Tables S4a and S4b).</p><p>In summary, we describe a successful approval of the MM MRD test by NGF as an FDA IDE, to be used as a decision-making tool for patient stratification in a clinical trial (packet content with submitted studies/files is listed in Table S5). Two main requirements by the FDA were (I) to show reliable MRD testing at a half-log higher sensitivity than the stated LOD of the assay; and (II) to show direct correlation of the MRD results with the FDA-approved NGS test (which had been approved in the time between our original CLIA-test validation and the time of IDE submission). We were able to achieve excellent correlation between NGF and NGS on a split sample cohort of specimens. Our experience may help other laboratories with IDE submissions and test validation, particularly in the new era of FDA regulation of LDTs. Furthermore, data presented here may be of interest to professional societies and companies seeking a higher level of FDA approval for the NGF in MM MRD testing (PMA or Premarket Notification 510(k)), as well as to individual clinical laboratories looking to validate MM MRD test as an LDT in the era of FDA oversight.</p><p>DJ, SKK, MS, and HO designed the study. DJ, MS, GEO, and HO analyzed the data. DJ wrote the draft of the letter. MS, PH, GEO, MMT, LBB, PTG, WIG, PK, MAG, MB, FKB, JZ, AD, TK, EM, SVM, SKK, and HO critically reviewed the data and the letter.</p><p>The authors declare no conflicts of interest.</p>","PeriodicalId":7724,"journal":{"name":"American Journal of Hematology","volume":"99 12","pages":"2399-2401"},"PeriodicalIF":10.1000,"publicationDate":"2024-09-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/ajh.27484","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"American Journal of Hematology","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/ajh.27484","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"HEMATOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Two recent decisions by the Food and Drug Administration will likely significantly impact testing for multiple myeloma (MM) minimal residual disease (MRD). First, on April 12, 2024, the FDA's Oncologic Drugs Advisory Committee (ODAC) voted to approve the use of MRD as an end point for accelerated approval of new treatments for patients with MM.1 This was a result of near 10 years effort by multiple institutions, professional societies, and patient's advocacy groups,2-6 and reflects the current state of MM treatment in which new therapeutic options have dramatically improved progression-free and overall survival (PFS and OS),7 making them impractical as the only clinical trial endpoints. Second, after many years of deliberations, FDA announced on April 29, 2024 that it will start overseeing laboratory developed tests (LDTs).8 LDTs are in vitro diagnostic products (IVDs) intended for clinical use; they are designed and validated for use within individual laboratories certified for performing high complexity testing under the Clinical Laboratory Improvement Amendments of 1988 (CLIA). The FDA's decision is currently being challenged by the American Clinical Laboratory Association.
The most commonly used assays for MRD detection in plasma cell neoplasms are next-generation sequencing (NGS) of the rearranged variable immunoglobulin heavy chain9, 10 and high sensitivity flow cytometry immunophenotyping (next-generation flow, NGF).11-14 Each type of assay has its advantages and disadvantages. For example, NGS requires knowledge of the diagnostic specimen for sequence comparison, but that knowledge enables higher sensitivity (10−6). NGF, as developed by the Euroflow/Spanish flow cytometry experts and commercialized by Cytognos requires no prior diagnostic specimen and can provide information regarding quality of the specimen (hemodilution); the sensitivity of the NGF is between 10−5 and 2 × 10−6, depending on the number of cells (events) collected. In 2016, the International Myeloma Working Group established the minimum sensitivity of 10−5 for MM MRD testing.4
Currently the only FDA-approved test for MM MRD is NGS-based clonoSEQ® by Adaptive Biotechnologies.9 For the NGF method to be FDA-approved, it will require a comprehensive package submission to the FDA, either as Premarket Approval (PMA) application or a Premarket Notification 510(k). However, FDA has a category of investigational device exemption (IDE) which allows a test to be used in a clinical study to collect safety and effectiveness data. There is limited data available regarding the requirements for FDA IDE approval of a flow cytometry assay for MRD testing.
In our laboratory, we adopted and validated the NGF method as an LDT, and have, since 2017, tested over 13 000 unique specimens (unpublished data). Our test validation protocol had been certified by CLIA and New York State Department of Health. However, when we submitted our validation documents as a part of the application for the FDA IDE for the BIQSFP-Funded Study EAA171, FDA requested additional validation experiments due to the fact that the MRD result was going to be used for MM patient stratification. After several meetings to precisely define additional requirements, we agreed with the FDA on the plan of action. Here we summarize additional experiments required by the FDA in order to approve IDE for the use of NGF in a clinical trial.
Analyte, specimen, and/or matrix stability: Additional four bone marrow samples were stored in shipping containers at ambient temperature and tested after 24, 48, 72, and 96 h. Results from the accepted time points had to be qualitatively the same (MRD-positive or negative). The last acceptable time point was defined as the one in which the coefficient of variation (CV) ≤25%. The data showed that under ambient shipping condition, MRD detection can be confidently assessed up to 96 h post-draw (Table S1).
Precision/reproducibility: Additional three MM samples were spiked into normal bone marrow samples, for calculated tumor burden between 5 × 10−6 and 5 × 10−5. A total of eight replicates were performed for each sample. CVs were calculated for comparisons of intra-assay, inter-assay, operator-to-operator, and instrument-to-instrument precision. Each replicate had to be qualitatively the same, with all the CVs ≤25%. Standardized machine settings and standardized gating strategy showed a highly precise assay near the assay's limit of detection (LOD) (Tables S2a and S2b).
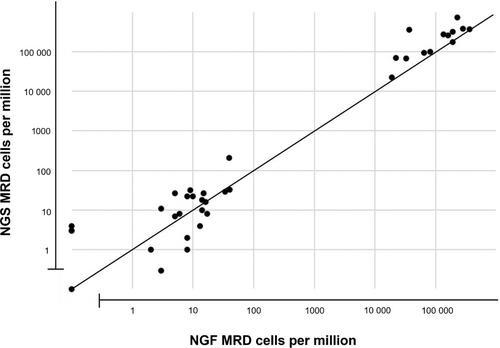
Accuracy: Twenty-one MM samples were used to compare MRD results by NGF with NGS-based clonoSEQ® assay, using split sample procedure. Three samples were tested with two different concentrations of spiked abnormal cells. Additional 13 diagnostic specimens were also a part of this comparison. The results showed 100% concordance of NGF with clonoSEQ® NGS testing for MRD ≥10−5. In addition, 10 out of 10 NGF-positive cases with the sensitivity between 2 × 10−6 and 10−5 were confirmed positive by NGS. Four NGF-negative cases showed very low level of positivity by NGS (3–4 × 10−6) (Figure 1 and Table S3). Overall Pearson correlation coefficient was 0.83.
Analytical sensitivity/limit of detection: Four MM bone marrow samples were spiked into normal bone marrow to achieve calculated tumor burden of 10−3 (baseline), 10−4, 10−5, and 5 × 10−6 (half-log below the assay LOD), in triplicates, with expected CVs ≤25%. Two samples showed precise MRD-positive LOD down to 2 × 10−6, and the other two down to 10−5 (Tables S4a and S4b).
In summary, we describe a successful approval of the MM MRD test by NGF as an FDA IDE, to be used as a decision-making tool for patient stratification in a clinical trial (packet content with submitted studies/files is listed in Table S5). Two main requirements by the FDA were (I) to show reliable MRD testing at a half-log higher sensitivity than the stated LOD of the assay; and (II) to show direct correlation of the MRD results with the FDA-approved NGS test (which had been approved in the time between our original CLIA-test validation and the time of IDE submission). We were able to achieve excellent correlation between NGF and NGS on a split sample cohort of specimens. Our experience may help other laboratories with IDE submissions and test validation, particularly in the new era of FDA regulation of LDTs. Furthermore, data presented here may be of interest to professional societies and companies seeking a higher level of FDA approval for the NGF in MM MRD testing (PMA or Premarket Notification 510(k)), as well as to individual clinical laboratories looking to validate MM MRD test as an LDT in the era of FDA oversight.
DJ, SKK, MS, and HO designed the study. DJ, MS, GEO, and HO analyzed the data. DJ wrote the draft of the letter. MS, PH, GEO, MMT, LBB, PTG, WIG, PK, MAG, MB, FKB, JZ, AD, TK, EM, SVM, SKK, and HO critically reviewed the data and the letter.
期刊介绍:
The American Journal of Hematology offers extensive coverage of experimental and clinical aspects of blood diseases in humans and animal models. The journal publishes original contributions in both non-malignant and malignant hematological diseases, encompassing clinical and basic studies in areas such as hemostasis, thrombosis, immunology, blood banking, and stem cell biology. Clinical translational reports highlighting innovative therapeutic approaches for the diagnosis and treatment of hematological diseases are actively encouraged.The American Journal of Hematology features regular original laboratory and clinical research articles, brief research reports, critical reviews, images in hematology, as well as letters and correspondence.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


