Capturing Correlation Effects in Positron Binding to Atoms and Molecules
IF 5.7
1区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
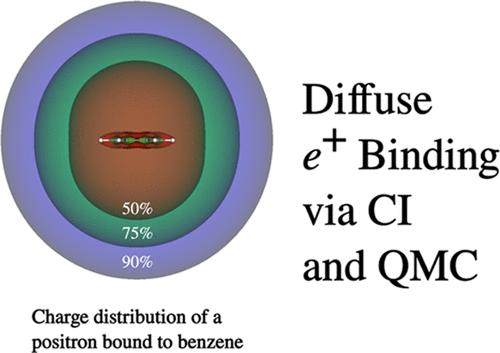
A major challenge in contemporary electronic structure theory involves the development of methods to describe in a balanced manner the contribution of correlation effects to energy differences. This challenge can be even greater for multicomponent systems containing more than one type of quantum particle. In the present work, we describe a flexible code for carrying out self-consistent field and configuration interaction (CI) calculations on multicomponent systems and use it to generate trial wave functions for use in diffusion Monte Carlo (DMC) calculations of the positron affinity of Be, Be2, Be4, Mg, CS2, and benzene. The resulting positron affinities (PAs) are in good agreement with the best values from the literature.
捕捉正电子与原子和分子结合的相关效应
当代电子结构理论面临的一个重大挑战是,如何开发出一种方法,以平衡的方式描述相关效应对能量差异的贡献。对于包含一种以上量子粒子的多组分系统来说,这一挑战可能更大。在本研究中,我们描述了一种灵活的代码,用于对多组分系统进行自洽场和构型相互作用(CI)计算,并利用它生成试验波函数,用于 Be、Be2、Be4、Mg、CS2 和苯的正电子亲和力的扩散蒙特卡罗(DMC)计算。计算得出的正电子亲和力 (PA) 与文献中的最佳值非常吻合。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of Chemical Theory and Computation
化学-物理:原子、分子和化学物理
CiteScore
9.90
自引率
16.40%
发文量
568
审稿时长
1 months
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


