The Persistence of Hydrogen Bonds in Pyrimidinones: From Solution to Crystal
IF 3.3
Q2 CHEMISTRY, MULTIDISCIPLINARY
引用次数: 0
Abstract
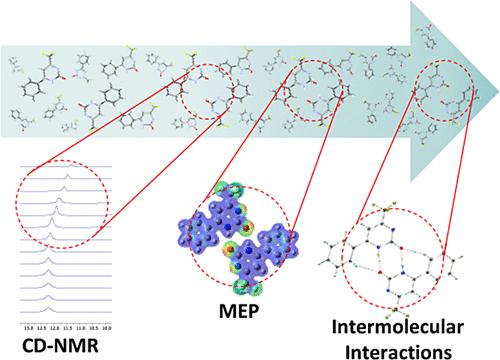
Pyrimidinone scaffolds are present in a wide array of molecules with synthetic and pharmacological utility. The inherent properties of these compounds may be attributed to intermolecular interactions analogous to the interactions that molecules tend to establish with active sites. Pyrimidinones and their fused derivatives have garnered significant interest due to their structural features, which resemble nitrogenous bases, the foundational building blocks of DNA and RNA. Similarly, pyrimidinones are predisposed to forming N–H···O hydrogen bonds akin to nitrogenous bases. Given this context, this study explored the supramolecular features and the predisposition to form hydrogen bonds in a series of 18 substituted 4-(trihalomethyl)-2(1H)-pyrimidinones. The formation of hydrogen bonds was observed in solution via nuclear magnetic resonance (NMR) spectroscopy experiments, and subsequently confirmed in the crystalline solid state. Hence, the 18 compounds were crystallized through crystallization assays by slow solvent evaporation, followed by single-crystal X-ray diffraction (SC-XRD). The supramolecular cluster demarcation was employed to evaluate all intermolecular interactions, and all crystalline structures exhibited robust hydrogen bonds, with an average energy of approximately −21.64 kcal mol–1 (∼19% of the total stabilization energy of the supramolecular clusters), irrespective of the substituents at positions 4, 5, or 6 of the pyrimidinone core. To elucidate the nature of these hydrogen bonds, an analysis based on the quantum theory of atoms in molecules (QTAIM) revealed that the predominant intermolecular interactions are N–H···O (average of −16.55 kcal mol–1) and C–H···O (average of −6.48 kcal mol–1). Through proposing crystallization mechanisms based on molecular stabilization energy data and contact areas between molecules and employing the supramolecular cluster and retrocrystallization concepts, it was determined that altering the halogen (F/Cl) at position 4 of the pyrimidinone nucleus modifies the crystallization mechanism pathway. Notably, the hydrogen bonds present in the initial proposed steps were confirmed by 1H NMR experiments using concentration-dependent techniques.
嘧啶酮中氢键的持久性:从溶液到晶体
嘧啶酮支架存在于多种具有合成和药理作用的分子中。这些化合物的固有特性可归因于分子间的相互作用,类似于分子与活性位点之间的相互作用。嘧啶酮类化合物及其融合衍生物因其结构特征类似于 DNA 和 RNA 的基本组成单元--含氮碱基而备受关注。同样,嘧啶酮也容易形成类似含氮碱基的 N-H-O 氢键。有鉴于此,本研究探讨了一系列 18 种取代的 4-(三卤甲基)-2(1H)-嘧啶酮的超分子特征和形成氢键的倾向。通过核磁共振(NMR)光谱实验在溶液中观察到了氢键的形成,随后在结晶固态中也得到了证实。因此,这 18 种化合物通过缓慢溶剂蒸发结晶试验结晶,然后进行单晶 X 射线衍射(SC-XRD)。所有结晶结构都显示出强大的氢键,其平均能量约为 -21.64 kcal mol-1(占超分子簇总稳定能的 19%),与嘧啶酮核心 4、5 或 6 位上的取代基无关。为了阐明这些氢键的性质,基于分子中原子量子理论(QTAIM)的分析表明,分子间的主要相互作用是 N-H--O(平均 -16.55 kcal mol-1)和 C-H--O(平均 -6.48 kcal mol-1)。根据分子稳定能数据和分子间接触面积提出结晶机制,并运用超分子簇和逆结晶概念,确定改变嘧啶酮核第 4 位的卤素(F/Cl)会改变结晶机制路径。值得注意的是,利用浓度依赖技术进行的 1H NMR 实验证实了最初提出的步骤中存在的氢键。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

ACS Organic & Inorganic Au
有机化学、无机化学-
CiteScore
4.10
自引率
0.00%
发文量
0
期刊介绍:
ACS Organic & Inorganic Au is an open access journal that publishes original experimental and theoretical/computational studies on organic organometallic inorganic crystal growth and engineering and organic process chemistry. Short letters comprehensive articles reviews and perspectives are welcome on topics that include:Organic chemistry Organometallic chemistry Inorganic Chemistry and Organic Process Chemistry.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


