Mechanistic Insights into Rh-Catalyzed Asymmetric Synthesis of Silicon-Stereogenic Silazanes: The Origin of Enantioselectivity
IF 2.5
3区 化学
Q2 CHEMISTRY, INORGANIC & NUCLEAR
引用次数: 0
Abstract
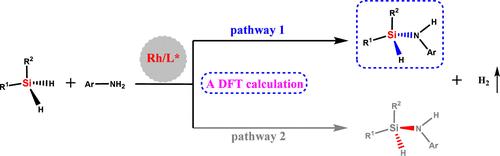
The catalytic asymmetric synthesis of silazanes is always a challenging task. Here, a highly enantioselective synthesis of silicon-stereogenic silazanes was investigated to elucidate the protocol’s principal features and to clarify the origin of the enantioselectivity by using DFT calculations. The computational results indicate that the total free energy barrier for the conversion is 19.9 kcal/mol, which is reasonable given the current reaction conditions. Consistent with the experimental findings, the calculations indicate that σ-bond metathesis (N–H bond cleavage) is the rate-determining step for this transformation. Both pathways 1 and 2 toward S- or R-configuration products were investigated computationally. We found that the main enantiomer product of this transformation is determined by the kinetically more favorable main reaction pathway 1. Calculations indicate that the loss of one or the other H on the dihydrosilane will lock the product chirality; therefore, the oxidative addition is the enantioselectivity-determining step. Non-covalent interaction (NCI) analysis confirms that a difference in steric hindrance is responsible for the enantioselectivity of the protocol. Additionally, calculations confirm that the electron-donating group on aniline appropriately lowers the free energy barrier relative to the electron-withdrawing group (ΔG = 15.5 vs 21.6 kcal/mol), thereby accelerating the conversion.
对 Rh 催化不对称合成硅烷的机理认识:不对称选择性的起源
硅烷的催化不对称合成一直是一项具有挑战性的任务。在此,我们利用 DFT 计算研究了硅稳定硅烷的高对映选择性合成,以阐明该方案的主要特征,并阐明对映选择性的来源。计算结果表明,在当前的反应条件下,转化的总自由能障为 19.9 kcal/mol,这是合理的。计算结果与实验结果一致,表明σ键元合成(N-H 键裂解)是这一转化的决定性步骤。我们通过计算研究了通向 S 或 R 构型产物的途径 1 和 2。我们发现,这种转化的主要对映体产物是由动力学上更有利的主要反应途径 1 决定的。计算表明,失去二氢硅烷上的一个或另一个 H 将锁定产物的手性;因此,氧化加成是决定对映体选择性的步骤。非共价相互作用(NCI)分析证实,立体阻碍的差异是该方案产生对映体选择性的原因。此外,计算还证实,苯胺上的供电子基团相对于抽电子基团适当地降低了自由能垒(ΔG = 15.5 vs 21.6 kcal/mol),从而加速了转化。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Organometallics
化学-无机化学与核化学
CiteScore
5.60
自引率
7.10%
发文量
382
审稿时长
1.7 months
期刊介绍:
Organometallics is the flagship journal of organometallic chemistry and records progress in one of the most active fields of science, bridging organic and inorganic chemistry. The journal publishes Articles, Communications, Reviews, and Tutorials (instructional overviews) that depict research on the synthesis, structure, bonding, chemical reactivity, and reaction mechanisms for a variety of applications, including catalyst design and catalytic processes; main-group, transition-metal, and lanthanide and actinide metal chemistry; synthetic aspects of polymer science and materials science; and bioorganometallic chemistry.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


