Analysis and Visualization of Quantitative Proteomics Data Using FragPipe-Analyst
IF 4.3
3区 材料科学
Q1 ENGINEERING, ELECTRICAL & ELECTRONIC
引用次数: 0
Abstract
The FragPipe computational proteomics platform is gaining widespread popularity among the proteomics research community because of its fast processing speed and user-friendly graphical interface. Although FragPipe produces well-formatted output tables that are ready for analysis, there is still a need for an easy-to-use and user-friendly downstream statistical analysis and visualization tool. FragPipe-Analyst addresses this need by providing an R shiny web server to assist FragPipe users in conducting downstream analyses of the resulting quantitative proteomics data. It supports major quantification workflows, including label-free quantification, tandem mass tags, and data-independent acquisition. FragPipe-Analyst offers a range of useful functionalities, such as various missing value imputation options, data quality control, unsupervised clustering, differential expression (DE) analysis using Limma, and gene ontology and pathway enrichment analysis using Enrichr. To support advanced analysis and customized visualizations, we also developed FragPipeAnalystR, an R package encompassing all FragPipe-Analyst functionalities that is extended to support site-specific analysis of post-translational modifications (PTMs). FragPipe-Analyst and FragPipeAnalystR are both open-source and freely available.
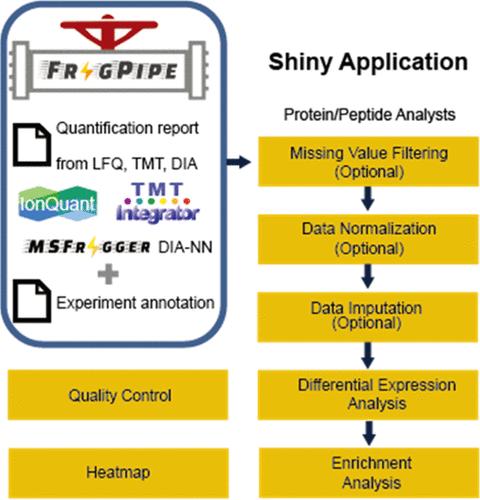
使用 FragPipe-Analyst 对定量蛋白质组学数据进行分析和可视化
FragPipe 计算蛋白质组学平台因其处理速度快和用户友好的图形界面而在蛋白质组学研究领域广受欢迎。尽管 FragPipe 能生成格式良好的输出表格以供分析,但仍需要一种易于使用、用户友好的下游统计分析和可视化工具。FragPipe-Analyst 通过提供一个闪亮的 R 网络服务器来满足这一需求,帮助 FragPipe 用户对生成的定量蛋白质组学数据进行下游分析。它支持主要的定量工作流程,包括无标记定量、串联质量标记和数据独立采集。FragPipe-Analyst 提供一系列有用的功能,如各种缺失值估算选项、数据质量控制、无监督聚类、使用 Limma 进行差异表达 (DE) 分析,以及使用 Enrichr 进行基因本体和通路富集分析。为了支持高级分析和定制可视化,我们还开发了 FragPipeAnalystR,这是一个包含 FragPipe-Analyst 所有功能的 R 软件包,可扩展到支持特定位点的翻译后修饰 (PTM) 分析。FragPipe-Analyst 和 FragPipeAnalystR 均开源并免费提供。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


