Eliminating redox-mediated electron transfer mechanisms on a supported molecular catalyst enables CO2 conversion to ethanol
IF 42.8
1区 化学
Q1 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
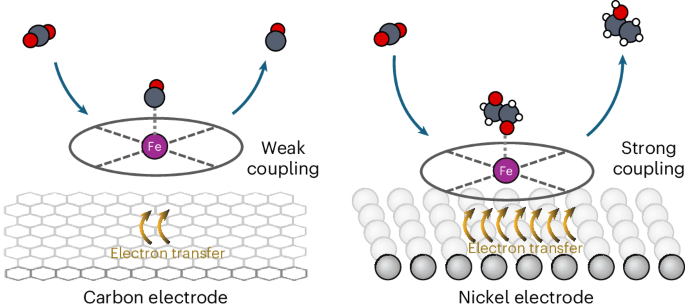
Molecular catalysts play a significant role in chemical transformations, utilizing changes in redox states to facilitate reactions. To date molecular electrocatalysts have efficiently produced single-carbon products from CO2 but have struggled to achieve a carbon–carbon coupling step. Conversely, copper catalysts can enable carbon–carbon coupling, but lead to broad C2+ product spectra. Here we subvert the traditional redox-mediated reaction mechanisms of organometallic compounds through a heterogeneous nickel-supported iron tetraphenylporphyrin electrocatalyst, facilitating electrochemical carbon–carbon coupling to produce ethanol. This represents a marked behavioural shift compared with carbon-supported metalloporphyrins. Extending the approach to a three-dimensional porous nickel support with adsorbed iron tetraphenylporphyrin, we attain ethanol Faradaic efficiencies of 68% ± 3.2% at −0.3 V versus a reversible hydrogen electrode (pH 7.7) with partial ethanol current densities of −21 mA cm−2. Separately we demonstrate maintained ethanol production over 60 h of operation. Further consideration of the wide parameter space of molecular catalyst and metal electrodes shows promise for additional chemistries and achievable metrics. The electrochemical reduction of CO2 on organometallic catalysts is commonly limited to two-electron products. Now, an iron tetraphenylporphyrin catalyst immobilized onto a nickel electrode is shown to achieve a Faradaic efficiency for ethanol of 68% due to the strong electronic coupling between the catalyst and the support.

消除支撑分子催化剂上氧化还原介导的电子传递机制,实现二氧化碳到乙醇的转化
分子催化剂利用氧化还原状态的变化促进反应,在化学转化中发挥着重要作用。迄今为止,分子电催化剂能有效地从二氧化碳中产生单碳产物,但却难以实现碳-碳偶联步骤。相反,铜催化剂可以实现碳-碳偶联,但会导致宽泛的 C2+ 产物光谱。在这里,我们通过一种异质镍支撑的四苯基卟啉铁电催化剂,颠覆了有机金属化合物传统的氧化还原介导反应机制,促进了电化学碳-碳偶联生成乙醇。与碳支撑金属卟啉相比,这是一种明显的行为转变。将这种方法扩展到吸附了四苯基卟啉铁的三维多孔镍载体上,我们在-0.3 V电压下与可逆氢电极(pH 值为 7.7)相比,乙醇法拉第效率达到 68% ± 3.2%,乙醇部分电流密度为 -21 mA cm-2。另外,我们还展示了运行 60 小时后乙醇的持续生产情况。对分子催化剂和金属电极的宽广参数空间的进一步研究表明,我们有望获得更多的化学成分和可实现的指标。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Nature Catalysis
Chemical Engineering-Bioengineering
CiteScore
52.10
自引率
1.10%
发文量
140
期刊介绍:
Nature Catalysis serves as a platform for researchers across chemistry and related fields, focusing on homogeneous catalysis, heterogeneous catalysis, and biocatalysts, encompassing both fundamental and applied studies. With a particular emphasis on advancing sustainable industries and processes, the journal provides comprehensive coverage of catalysis research, appealing to scientists, engineers, and researchers in academia and industry.
Maintaining the high standards of the Nature brand, Nature Catalysis boasts a dedicated team of professional editors, rigorous peer-review processes, and swift publication times, ensuring editorial independence and quality. The journal publishes work spanning heterogeneous catalysis, homogeneous catalysis, and biocatalysis, covering areas such as catalytic synthesis, mechanisms, characterization, computational studies, nanoparticle catalysis, electrocatalysis, photocatalysis, environmental catalysis, asymmetric catalysis, and various forms of organocatalysis.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


