Jonathan Donhauser , Anna Doménech-Pascual , Xingguo Han , Karen Jordaan , Jean-Baptiste Ramond , Aline Frossard , Anna M. Romaní , Anders Priemé
{"title":"Modelling soil prokaryotic traits across environments with the trait sequence database ampliconTraits and the R package MicEnvMod","authors":"Jonathan Donhauser , Anna Doménech-Pascual , Xingguo Han , Karen Jordaan , Jean-Baptiste Ramond , Aline Frossard , Anna M. Romaní , Anders Priemé","doi":"10.1016/j.ecoinf.2024.102817","DOIUrl":null,"url":null,"abstract":"<div><p>We present a comprehensive, customizable workflow for inferring prokaryotic phenotypic traits from marker gene sequences and modelling the relationships between these traits and environmental factors, thus overcoming the limited ecological interpretability of marker gene sequencing data. We created the trait sequence database <em>ampliconTraits</em>, constructed by cross-mapping species from a phenotypic trait database to the SILVA sequence database and formatted to enable seamless classification of environmental sequences using the SINAPS algorithm. The R package <em>MicEnvMod</em> enables modelling of trait – environment relationships, combining the strengths of different model types and integrating an approach to evaluate the models' predictive performance in a single framework. Traits could be accurately predicted even for sequences with low sequence identity (80 %) with the reference sequences, indicating that our approach is suitable to classify a wide range of environmental sequences. Validating our approach in a large trans-continental soil dataset, we showed that trait distributions were robust to classification settings such as the bootstrap cutoff for classification and the number of discrete intervals for continuous traits. Using functions from <em>MicEnvMod,</em> we revealed precipitation seasonality and land cover as the most important predictors of genome size. We found Pearson correlation coefficients between observed and predicted values up to 0.70 using repeated split sampling cross validation, corroborating the predictive ability of our models beyond the training data. Predicting genome size across the Iberian Peninsula, we found the largest genomes in the northern part. Potential limitations of our trait inference approach include dependence on the phylogenetic conservation of traits and limited database coverage of environmental prokaryotes. Overall, our approach enables robust inference of ecologically interpretable traits combined with environmental modelling allowing to harness traits as bioindicators of soil ecosystem functioning.</p></div>","PeriodicalId":51024,"journal":{"name":"Ecological Informatics","volume":"83 ","pages":"Article 102817"},"PeriodicalIF":5.8000,"publicationDate":"2024-09-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S1574954124003595/pdfft?md5=a975351ee65c86e764ade9d9b4d869ae&pid=1-s2.0-S1574954124003595-main.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Ecological Informatics","FirstCategoryId":"93","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1574954124003595","RegionNum":2,"RegionCategory":"环境科学与生态学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"ECOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
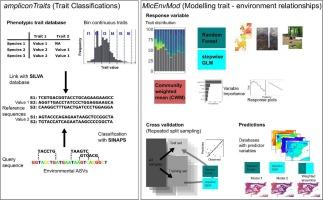
We present a comprehensive, customizable workflow for inferring prokaryotic phenotypic traits from marker gene sequences and modelling the relationships between these traits and environmental factors, thus overcoming the limited ecological interpretability of marker gene sequencing data. We created the trait sequence database ampliconTraits, constructed by cross-mapping species from a phenotypic trait database to the SILVA sequence database and formatted to enable seamless classification of environmental sequences using the SINAPS algorithm. The R package MicEnvMod enables modelling of trait – environment relationships, combining the strengths of different model types and integrating an approach to evaluate the models' predictive performance in a single framework. Traits could be accurately predicted even for sequences with low sequence identity (80 %) with the reference sequences, indicating that our approach is suitable to classify a wide range of environmental sequences. Validating our approach in a large trans-continental soil dataset, we showed that trait distributions were robust to classification settings such as the bootstrap cutoff for classification and the number of discrete intervals for continuous traits. Using functions from MicEnvMod, we revealed precipitation seasonality and land cover as the most important predictors of genome size. We found Pearson correlation coefficients between observed and predicted values up to 0.70 using repeated split sampling cross validation, corroborating the predictive ability of our models beyond the training data. Predicting genome size across the Iberian Peninsula, we found the largest genomes in the northern part. Potential limitations of our trait inference approach include dependence on the phylogenetic conservation of traits and limited database coverage of environmental prokaryotes. Overall, our approach enables robust inference of ecologically interpretable traits combined with environmental modelling allowing to harness traits as bioindicators of soil ecosystem functioning.
期刊介绍:
The journal Ecological Informatics is devoted to the publication of high quality, peer-reviewed articles on all aspects of computational ecology, data science and biogeography. The scope of the journal takes into account the data-intensive nature of ecology, the growing capacity of information technology to access, harness and leverage complex data as well as the critical need for informing sustainable management in view of global environmental and climate change.
The nature of the journal is interdisciplinary at the crossover between ecology and informatics. It focuses on novel concepts and techniques for image- and genome-based monitoring and interpretation, sensor- and multimedia-based data acquisition, internet-based data archiving and sharing, data assimilation, modelling and prediction of ecological data.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


