Internal Conversion Cascade in a Carbon Nanobelt: A Multiconfigurational Quantum Dynamical Study
IF 5.5
1区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
Carbon nanobelts feature intriguing photophysical properties, due to their high symmetry and structural rigidity. Here, we consider a (6,6) armchair carbon nanobelt, i.e., the very first carbon nanobelt to be synthesized [Povie et al., Science 2017, 356, 172] and characterize the internal conversion dynamics using multiconfigurational quantum dynamics via the multi-layer multiconfiguration time-dependent Hartree (ML-MCTDH) method. A symmetry-adapted linear vibronic coupling Hamiltonian for 26 electronic states and 210 vibrational modes is employed. Electronic excitations are found to decay through a dense manifold of excited states, which interact via multiple conical intersections, while inducing minimal geometry change. It is shown that a rapid coherent decay, exhibiting a nonvanishing quantum flux on a time scale of less than 50 fs, transitions toward a slower, decoherent decay at longer times. As previously suggested in the literature, electronic relaxation is hindered by phonon bottlenecks such that a stepwise internal conversion cascade is observed. The computed vibronic absorption spectrum is shown to be in good agreement with the experimental spectrum.
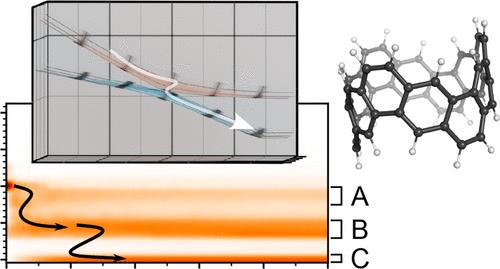
碳纳米带中的内部转换级联:多构型量子动力学研究
碳纳米带因其高度对称性和结构刚性而具有引人入胜的光物理特性。在此,我们考虑了 (6,6) 扶手碳纳米带,即第一个被合成的碳纳米带 [Povie 等人,Science 2017, 356, 172],并通过多层多配置时间相关哈特里(ML-MCTDH)方法,利用多配置量子动力学表征了内部转换动力学。该方法采用了对称调整的线性振子耦合哈密顿,包含 26 个电子态和 210 个振动模式。研究发现,电子激发通过密集的激发态流形衰减,这些激发态通过多个锥形交叉点相互作用,同时引起最小的几何变化。研究表明,快速相干衰变在小于 50 fs 的时间尺度上表现出不消失的量子通量,而在更长的时间内则过渡到较慢的非相干衰变。正如以前的文献所指出的,电子弛豫受到声子瓶颈的阻碍,因此观察到一种逐步的内部转换级联。计算得出的振动吸收光谱与实验光谱十分吻合。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of Chemical Theory and Computation
化学-物理:原子、分子和化学物理
CiteScore
9.90
自引率
16.40%
发文量
568
审稿时长
1 months
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


